https://github.com/OpenGene/MutScan
Detect and visualize target mutations by scanning FastQ files directly
https://github.com/OpenGene/MutScan
bioinformatics cancer detection fastq mutation ngs somatic validation variant visualization
Last synced: about 1 year ago
JSON representation
Detect and visualize target mutations by scanning FastQ files directly
- Host: GitHub
- URL: https://github.com/OpenGene/MutScan
- Owner: OpenGene
- License: mit
- Created: 2016-07-23T02:34:15.000Z (about 10 years ago)
- Default Branch: master
- Last Pushed: 2022-02-10T01:52:44.000Z (over 4 years ago)
- Last Synced: 2025-04-10T09:54:06.789Z (over 1 year ago)
- Topics: bioinformatics, cancer, detection, fastq, mutation, ngs, somatic, validation, variant, visualization
- Language: C
- Homepage:
- Size: 914 KB
- Stars: 151
- Watchers: 21
- Forks: 39
- Open Issues: 6
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
- top-life-sciences - **OpenGene/MutScan** - 02-10 01:52:44 | (Ranked by starred repositories)
README
[](https://anaconda.org/bioconda/mutscan)
# MutScan
Detect and visualize target mutations by scanning FastQ files directly
* [Features](#features)
* [Application scenarios](#application-scenarios)
* [Take a quick glance](#take-a-quick-glance)
* [Download, compile and install](#get-mutscan)
* [HTML report](#html-report)
* [JSON report](#json-report)
* [All options](#all-options)
* [Customize your mutation file](#mutation-file)
* [Work with BAM/CRAM](#work-with-bamcram)
* [Remarks](#remarks)
* [Cite MutScan](#cite-mutscan)
# Features
* Ultra sensitive, guarantee that all reads supporting the mutations will be detected
* Can be 50X+ faster than normal pipeline (i.e. BWA + Samtools + GATK/VarScan/Mutect).
* Very easy to use and need nothing else. No alignment, no reference genome, no variant call, no...
* Contains built-in most actionable mutation points for cancer-related mutations, like EGFR p.L858R, BRAF p.V600E...
* Beautiful and informative HTML report with informative pileup visualization.
* Multi-threading support.
* Supports both single-end and pair-end data.
* For pair-end data, MutScan will try to merge each pair, and do quality adjustment and error correction.
* Able to scan the mutations in a VCF file, which can be used to visualize called variants.
* Can be used to filter false-positive mutations. i.e. MutScan can handle highly repetive sequence to avoid false INDEL calling.
# Application scenarios:
* you are interested in some certain mutations (like cancer drugable mutations), and want to check whether the given FastQ files contain them.
* you have no enough confidence with the mutations called by your pipeline, so you want to visualize and validate them to avoid false positive calling.
* you worry that your pipeline uses too strict filtering and may cause some false negative, so you want to check that in a fast way.
* you want to visualize the called mutation and take a screenshot with its clear pipeup information.
* you called a lot of INDEL mutations, and you worry that mainly they are false positives (especially in highly repetive region)
* you want to validate and visualize every record in the VCF called by your pipeline.
* ...
# Take a quick glance
* Sample HTML report: http://opengene.org/MutScan/report.html
* Sample JSON report: http://opengene.org/MutScan/report.json
* Dataset for testing: http://opengene.org/dataset.html
* Command to test
```shell
mutscan -1 R1.fq.gz -2 R2.fq.gz
```
# Get MutScan
## install with Bioconda
[](https://anaconda.org/bioconda/mutscan)
```shell
conda install -c bioconda mutscan
```
## download binary
This binary is only for Linux systems: http://opengene.org/MutScan/mutscan
```shell
# this binary was compiled on CentOS, and tested on CentOS/Ubuntu
wget http://opengene.org/MutScan/mutscan
chmod a+x ./mutscan
```
## or compile from source
```shell
# get source (you can also use browser to download from master or releases)
git clone https://github.com/OpenGene/MutScan.git
# build
cd mutscan
make
# Install
sudo make install
```
# Windows version (may be not the latest version)
If you want to compile MutScan on Windows, you should use `cygwin`. We already built one with cygwin-2.6.0/g++ 5.4, and it can be downloaded from:
http://opengene.org/MutScan/windows_mutscan.zip
# HTML report
* A HTML report will be generated, and written to the given filename. See http://opengene.org/MutScan/report.html for an example.
* ***If you run the command in your Linux server and want to view the HTML report on your local system. DO remember to copy all of the `xxxx.html` and `xxxx.html.files` and keep them in the same folder, then click `xxxx.html` to view it in browser.***
* The default file name is `mutscan.html`, and a folder `mutscan.html.files` will be also generated.
* By default, an indivudal HTML file will be generated for each found mutation. But you can specify `-s` or `--standalone` to contain all mutations in a single HTML file. Be caution with this mode if you are scanning too many records (for example, scanning VCF), it will give you a very big HTML file and is not loadable by browser.
* Here is a screenshot for the pileup of a mutation (EGFR p.T790M) generated by MutScan:
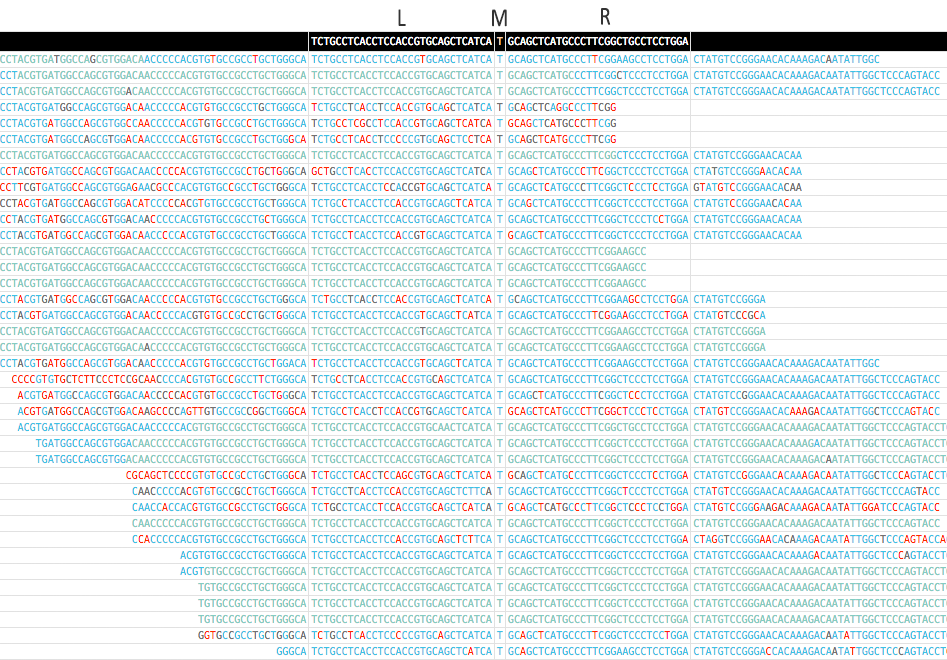
* An pileup of EGFR p.T790M mutation is displayed above. EGFR p.T790M is a very important drugable mutation for lung cancer.
* The color of each base indicates its quality, and the quality will be shown when mouse over.
* In first column, d means the edit distance of match, and --> means forward, <-- means reverse
# JSON report
JSON report is disabled by default. You can enable it by specifying a JSON file name using `-j` or `--json`. A JSON report is like this:
```json
{
"command":"./mutscan -1 /Users/shifu/data/fq/S010_20170320003-4_ffpedna_pan-cancer-v1_S10_R1_001.fastq -2 /Users/shifu/data/fq/S010_20170320003-4_ffpedna_pan-cancer-v1_S10_R2_001.fastq -h z.html -j z.json -v --simplified=off ",
"version":"1.14.0",
"time":"2018-05-15 15:48:21",
"mutations":{
"NRAS-neg-1-115258747-2-c.35G>C-p.G12A-COSM565":{
"chr":"chr1",
"ref":["TGGATTGTCAGTGCGCTTTTCCCAACACCA","G","CTGCTCCAACCACCACCAGTTTGTACTCAG"],
"reads":[
{
"breaks":[31,61,62,76],
"seq":"ATATTCATCTACAAAGTGGTTCTGGATTAGCTGGATTGTCAGTGCGCTTTTCCCAACACCAGCTGCTCCAACCACC",
"qual":"eeeeeiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiieieeeee"
},
{
"breaks":[31,61,62,76],
"seq":"ATATTCATCTACAAAGTGGTTCTGGATTAGCTGGATTGTCAGTGCGCTTTTCCCAACACCAGCTGCTCCAACCACC",
"qual":"eeeeeiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieeeee"
}
]
},
"PIK3CA-pos-3-178936082-9-c.1624G>A-E542K-COSM760":{
"chr":"chr3",
"ref":["AAAGCAATTTCTACACGAGATCCTCTCTCT","A","AAATCACTGAGCAGGAGAAAGATTTTCTAT"],
"reads":[
{
"breaks":[22,52,53,83],
"seq":"GGAAAATGACAAAGAACAGCTCAAAGCAATTTCTACACGAGATCCTCTCTCTAAAATCACTGAGCAGGAGAAAGATTTTCCAAAGATGTTTCTCAGAACGCTGCAGTCTGCAATTTGTATGAATTCCC",
"qual":"eeeeeiiiQiiiiiieiiiieiSeiiiiiie`iiii`i`iiiiiiiiiiiiii`iiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiaiiiiiiiiiiiiiiiiiieiiiiiieeeee"
},
{
"breaks":[0,27,28,58],
"seq":"GCAATTTCTACACGAGATCCTCTCTCTAAAATCACTGCGCAGGAGAAAGATTTTCTATGGACCACAGGTAAGTGCTAAAATGGAGATTCTCTGTTTCTTTTTCTTTATTACAGAAAAAATAACTGACTTTGGCTGATCTCAGCATGTTTTTACCATACC",
"qual":"AAAAAEEEEiieiiieiiiiiiiiiieiiiiiiiie``iiiiiieiiiiiiiiiieiiiieiieieeiiiSiiiiiieiiiiiiiiiiiiiieiiiiiSiiiiiiiiiiiiieiiiiiiiiiiii`ieiiieiii`ieiiiii`eS``eieEEEAAAAA"
}
]
}
}
}
```
# All options
```shell
usage: mutscan -1 -2 [options]...
options:
-1, --read1 read1 file name, required
-2, --read2 read2 file name
-m, --mutation mutation file name, can be a CSV format or a VCF format
-r, --ref reference fasta file name (only needed when mutation file is a VCF)
-h, --html filename of html report, default is mutscan.html in work directory
-j, --json filename of JSON report, default is no JSON report (string [=])
-t, --thread worker thread number, default is 4
-S, --support min read support required to report a mutation, default is 2.
-k, --mark when mutation file is a vcf file, --mark means only process the records with FILTER column is M
-l, --legacy use legacy mode, usually much slower but may be able to find a little more reads in certain case
-s, --standalone output standalone HTML report with single file. Don't use this option when scanning too many target mutations (i.e. >1000 mutations)
-n, --no-original-reads dont output original reads in HTML and text output. Will make HTML report files a bit smaller
-?, --help print this message
```
The plain text result, which contains the detected mutations and their support reads, will be printed directly. You can use `>` to redirect output to a file, like:
```shell
mutscan -1 -2 > result.txt
```
MutScan generate a very informative HTML file report, default is `mutscan.html` in the work directory. You can change the file name with `-h` argument, like:
```
mutscan -1 -2 -h report.html
```
## single-end and pair-end
For single-end sequencing data, `-2` argument is omitted:
```
mutscan -1
```
## multi-threading
`-t` argument specify how many worker threads will be launched. The default thread number is `4`. Suggest to use a number less than the CPU cores of your system.
# Mutation file
* Mutation file, specified by `-m`, can be a `CSV file`, or a `VCF file`.
* If no `-m` specified, MutScan will use the built-in default mutation file with about 60 cancer related mutation points.
* If a CSV is provided, no reference genome assembly needed.
* If a VCF is provided, corresponding reference genome assembly should be provided (i.e. ucsc.hg19.fasta), and should not be zipped.
## CSV-format mutation file
A CSV file with columns of `name`, `left_seq_of_mutation_point`, `mutation_seq`, `right_seq_of_mutation_point` and `chromosome(optional)`
```csv
#name, left_seq_of_mutation_point, mutation_seq, right_seq_of_mutation_point, chromosome
NRAS-neg-1-115258748-2-c.34G>A-p.G12S-COSM563, GGATTGTCAGTGCGCTTTTCCCAACACCAC, T, TGCTCCAACCACCACCAGTTTGTACTCAGT, chr1
NRAS-neg-1-115252203-2-c.437C>T-p.A146V-COSM4170228, TGAAAGCTGTACCATACCTGTCTGGTCTTG, A, CTGAGGTTTCAATGAATGGAATCCCGTAAC, chr1
BRAF-neg-7-140453136-15-c.1799T>A -V600E-COSM476, AACTGATGGGACCCACTCCATCGAGATTTC, T, CTGTAGCTAGACCAAAATCACCTATTTTTA, chr7
EGFR-pos-7-55241677-18-c.2125G>A-p.E709K-COSM12988, CCCAACCAAGCTCTCTTGAGGATCTTGAAG, A, AAACTGAATTCAAAAAGATCAAAGTGCTGG, chr7
EGFR-pos-7-55241707-18-c.2155G>A-p.G719S-COSM6252, GAAACTGAATTCAAAAAGATCAAAGTGCTG, A, GCTCCGGTGCGTTCGGCACGGTGTATAAGG, chr7
EGFR-pos-7-55241707-18-c.2155G>T-p.G719C-COSM6253, GAAACTGAATTCAAAAAGATCAAAGTGCTG, T, GCTCCGGTGCGTTCGGCACGGTGTATAAGG, chr7
```
`testdata/mutations.csv` gives an example of CSV-format mutation file
## VCF-format mutation file
A standard VCF can be used as a mutation file, with file extension `.vcf` or `.VCF`. If the mutation file is a VCF file, you should specify the `reference assembly file` by `-r `. For example the command can be:
```shell
mutscan -1 R1.fq -2 R2.fq -m target.vcf -r hg19.fa
```
# Work with BAM/CRAM
If you want to run MutScan with BAM/CRAM files, you can use `samtools` to convert them to FASTQ files using `samtools fastq` command, both single-end and paired-end data are supported by latest version of `samtools fastq`.
# Remarks
* `MutScan` requires at least 50 bp long reads, if your reads are too short, do not use it
* If you want to extract mutations even with only one read support, add `-S 1` or `--support=1` in the command
* Feel free to raise an issue if you meet any problem
# Cite MutScan
Shifu Chen, Tanxiao Huang, TieXiang Wen, Hong Li, Mingyan Xu and Jia Gu. MutScan: fast detection and visualization of target mutations by scanning FASTQ data. BMC Bioinformatics. https://doi.org/10.1186/s12859-018-2024-6