https://github.com/ahwagner/pymvld
A Python implementation of the Minimum Variant Level Data standard
https://github.com/ahwagner/pymvld
genomics standards variant-interpretation
Last synced: about 1 year ago
JSON representation
A Python implementation of the Minimum Variant Level Data standard
- Host: GitHub
- URL: https://github.com/ahwagner/pymvld
- Owner: ahwagner
- License: mit
- Created: 2018-07-31T04:59:15.000Z (almost 8 years ago)
- Default Branch: master
- Last Pushed: 2018-08-25T05:36:38.000Z (almost 8 years ago)
- Last Synced: 2025-01-28T14:47:55.036Z (over 1 year ago)
- Topics: genomics, standards, variant-interpretation
- Language: Python
- Homepage:
- Size: 14.6 KB
- Stars: 0
- Watchers: 2
- Forks: 0
- Open Issues: 0
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
README
# pyMVLD
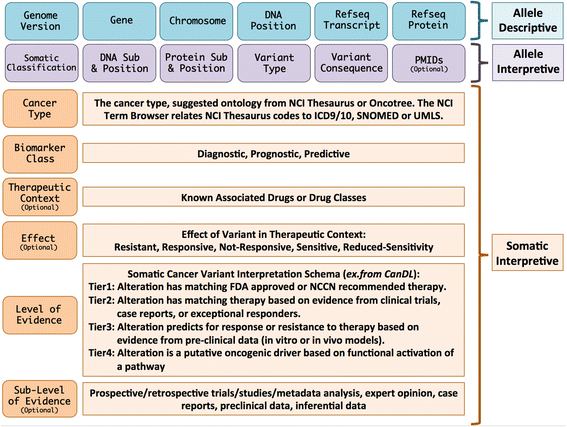
pyMVLD is a Python implementation of the Minimum Variant Level Data framework of standardized data elements, first defined by the ClinGen Somatic Working Group in 2016 ([paper](https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-016-0367-z)).
## Why use pyMVLD
pyMVLD is an implementation of the MVLD framework for ensuring compliance. Use of this module provides:
* Validation of correct data types and values when generating MVLD objects
* Immutable, standardized objects for downstream applications
* Framework versioning
## How to use pyMVLD
Creating an MVLD object first requires construction of three sub-objects corresponding to the three field sets described by the framework: _AlleleDescriptive_, _AlleleInterpretive_, and _SomaticInterpretive_.
### Allele Descriptive Fields
The Allele Descriptive fields are expected to conform to the following rules:
#### Genome Version
The Genome Version must be in the GRCh37/GRCh38 format, preferably with the build version (e.g. GRCh38.p12). pyMVLD explicitly limits the values to a `str` describing GRCh37 or GRCh38, the two major GRC assemblies that are currently available. The assembly version, if provided, is checked only for syntactic correctness, not that it corresponds to a published version.
#### Gene
Genes must be provided as HGNC Approved Gene Symbols. The `Gene` field is checked to be a `str`, and is compared against the current HGNC list of approved symbols. If not HGNC Approved, the raised error describes the use of a known alias or retired symbol, if applicable.
### Examples
```
kwargs = {
'genome_version': 'GRCh37',
'gene': 'BRAF',
'chromosome': 'chr7',
'dna_position': 'NC_000007.13:g.140453136A>T',
'refseq_transcript': 'NM_004333.4',
'refseq_protein': 'NP_004324.2'
}
ad = AlleleDescriptive(**kwargs)
```