https://github.com/cafferychen777/flashdeconv
Fast Linear Algebra for Scalable Hybrid Deconvolution of Spatial Transcriptomics
https://github.com/cafferychen777/flashdeconv
bioinformatics cell-type-deconvolution computational-biology deconvolution python scanpy single-cell spatial-transcriptomics visium
Last synced: 5 months ago
JSON representation
Fast Linear Algebra for Scalable Hybrid Deconvolution of Spatial Transcriptomics
- Host: GitHub
- URL: https://github.com/cafferychen777/flashdeconv
- Owner: cafferychen777
- License: bsd-3-clause
- Created: 2025-12-12T05:20:58.000Z (6 months ago)
- Default Branch: main
- Last Pushed: 2026-01-04T13:01:57.000Z (5 months ago)
- Last Synced: 2026-01-04T23:15:59.863Z (5 months ago)
- Topics: bioinformatics, cell-type-deconvolution, computational-biology, deconvolution, python, scanpy, single-cell, spatial-transcriptomics, visium
- Language: Python
- Size: 6.32 MB
- Stars: 3
- Watchers: 0
- Forks: 0
- Open Issues: 1
-
Metadata Files:
- Readme: README.md
- License: LICENSE
- Citation: CITATION.cff
Awesome Lists containing this project
- awesome-biological-image-analysis - FlashDeconv - High-performance spatial transcriptomics deconvolution for cell type mapping using structure-preserving randomized sketching. (Image processing and segmentation)
- awesome-computational-biology - FlashDeconv - High-performance spatial transcriptomics deconvolution. Processes 1M spots in ~3 minutes. (Preprocess / Clinical Trial)
- awesome-multi-omics - FlashDeconv - Yang - High-performance spatial transcriptomics deconvolution using structure-preserving sketching, processes 1M spots in ~3 min - [paper](https://doi.org/10.64898/2025.12.22.696108) (Software packages and methods / Single cell multi-omics)
- awesome-single-cell - FlashDeconv - [Python] A high-performance spatial transcriptomics deconvolution method using structure-preserving randomized sketching for atlas-scale and subcellular-resolution platforms. Processes 1 million spots in ~3 minutes with linear O(N) scaling. manuscript: [FlashDeconv enables atlas-scale, multi-resolution spatial deconvolution via structure-preserving sketching](https://doi.org/10.64898/2025.12.22.696108) (Software packages / Spatial transcriptomics)
README
# FlashDeconv
**Fast Linear Algebra for Scalable Hybrid Deconvolution**
[](https://pypi.org/project/flashdeconv/)
[](https://opensource.org/licenses/BSD-3-Clause)
[](https://www.python.org/downloads/)
[](https://doi.org/10.5281/zenodo.18109003)
*Unlocking atlas-scale spatial biology with randomized numerical linear algebra.*
FlashDeconv is a high-performance spatial transcriptomics deconvolution method designed for **atlas-scale** and **subcellular-resolution** platforms (Visium HD, Stereo-seq, Xenium). It leverages structure-preserving randomized sketching to estimate cell type proportions with linear scalability—processing **1 million spots in ~3 minutes** on commodity hardware.
> **Paper:** Chen Yang, Jun Chen, Xianyang Zhang. *FlashDeconv enables atlas-scale, multi-resolution spatial deconvolution via structure-preserving sketching*. bioRxiv, 2025. DOI: [10.64898/2025.12.22.696108](https://doi.org/10.64898/2025.12.22.696108)
>
> **Reproducibility:** To reproduce figures and benchmarks from the paper, visit the [flashdeconv-reproducibility](https://github.com/cafferychen777/flashdeconv-reproducibility) repository.
---
## Key Features
- **Ultra-fast & Scalable:** Deconvolve **1 million spots in ~3 minutes**. Time and memory scale linearly O(N) with dataset size.
- **Hardware Friendly:** No GPU required. Runs efficiently on laptops (e.g., 32GB RAM handles 1M spots).
- **Rare Cell Detection:** Uses **leverage-score sampling** to preserve transcriptomically distinct but low-abundance cell types (e.g., Tuft cells, endothelial cells) that variance-based methods systematically miss.
- **Spatially Aware:** Sparse graph Laplacian regularization ensures spatial coherence without the O(N²) cost of dense kernel methods.
- **Visium HD Ready:** Specifically optimized for the extreme sparsity and scale of subcellular resolution technologies (2µm–16µm bin sizes).
- **Statistically Rigorous:** Log-CPM normalization with leverage-weighted gene selection preserves both common and rare cell populations.
---
## Installation
```bash
# From PyPI (recommended)
pip install flashdeconv
# With scanpy/anndata integration
pip install flashdeconv[io]
```
**For development:**
```bash
# From source
git clone https://github.com/cafferychen777/flashdeconv.git
cd flashdeconv
pip install -e ".[dev]"
```
**Requirements:** Python ≥ 3.9, numpy, scipy, numba. Optional: scanpy, anndata for AnnData workflow.
---
## Quick Start
### With Scanpy/AnnData
```python
import scanpy as sc
import flashdeconv as fd
adata_st = sc.read_h5ad("visium_hd.h5ad")
adata_ref = sc.read_h5ad("reference.h5ad")
fd.tl.deconvolve(adata_st, adata_ref, cell_type_key="cell_type")
adata_st.obsm["flashdeconv"] # Cell type proportions
sc.pl.spatial(adata_st, color="flashdeconv_Hepatocyte")
```
### With NumPy
```python
from flashdeconv import FlashDeconv
model = FlashDeconv(lambda_spatial=5000)
proportions = model.fit_transform(Y, X, coords) # (n_spots, n_cell_types)
```
---
## Best Practices: Tuning `lambda_spatial`
While FlashDeconv works well with defaults, **adjusting `lambda_spatial`** (spatial regularization strength) based on your platform's **spot size** and **counts-per-spot** significantly improves results.
| Platform | Spot Size | Typical UMI/Spot | Recommended `lambda_spatial` | Rationale |
|:---------|:----------|:-----------------|:----------------------------|:----------|
| **Standard Visium** | 55µm | 10,000–30,000 | `1000–10000` (default: 5000) | Strong signal; minimal smoothing needed |
| **Visium HD (16µm)** | 16µm | 200–2,000 | `5000–20000` | Moderate sparsity; leverage neighbors |
| **Visium HD (8µm)** | 8µm | 50–500 | `10000–50000` | Very sparse; rely on spatial priors |
| **Visium HD (2µm)** | 2µm | 1–10 | `50000–100000` | Extreme sparsity; heavy smoothing |
| **Stereo-seq / Seq-Scope** | 0.5–1µm | 5–50 | `50000–200000` | Single-cell/subcellular resolution; extreme sparsity |
> **Note:**
> - If cell type maps look **"salt-and-pepper" noisy**, increase `lambda_spatial`
> - If maps look **overly blurred**, decrease `lambda_spatial`
> - Use `lambda_spatial="auto"` for automatic tuning (may underestimate for real data; best for initial exploration)
> - For **non-grid layouts** (e.g., Xenium, MERFISH), set `spatial_method="knn"` (default)
---
## Algorithm Under the Hood
FlashDeconv reformulates spatial deconvolution as **Graph-Regularized Non-Negative Least Squares**, solved in a compressed "sketch" space via randomized numerical linear algebra (RandNLA):
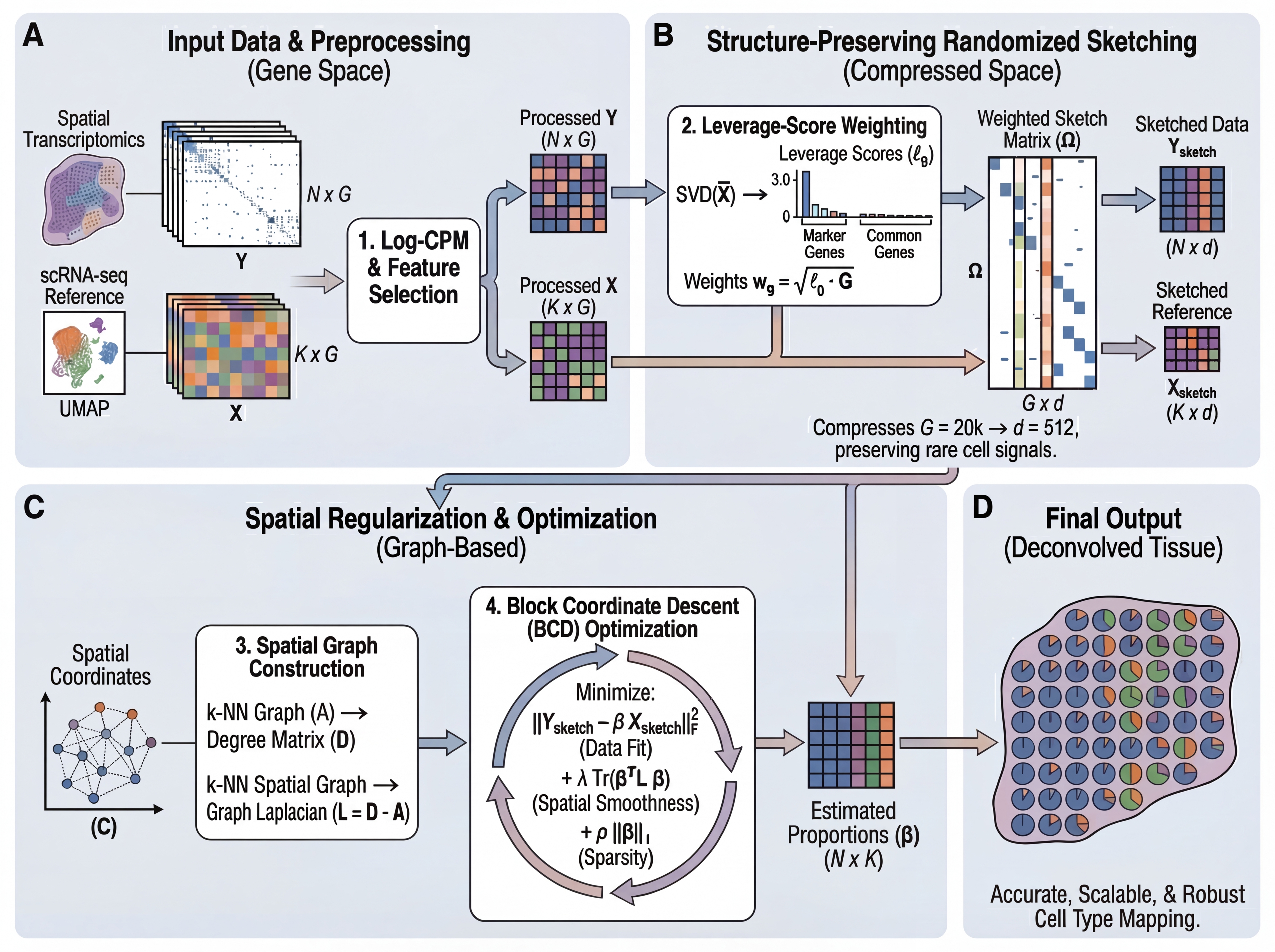
**Figure 1. Overview of the FlashDeconv framework.** (A) Input data preprocessing with Log-CPM normalization and gene selection. (B) Structure-preserving randomized sketching using leverage-score weighting to compress gene space while preserving rare cell signals. (C) Spatial graph construction and regularized optimization via Block Coordinate Descent. (D) Final cell type proportion estimates for each spatial location.
### Three-Stage Framework
1. **Preprocessing & Gene Selection**
- **Log-CPM normalization**: Stabilizes variance and prevents high-expression genes from dominating the sketch
- **Leverage-weighted gene selection**: Combines highly variable genes (HVGs) with cell-type-specific markers, weighted by statistical leverage scores. Unlike variance (which conflates abundance with informativeness), leverage scores identify genes that define **transcriptomically distinct directions**, preserving rare cell type markers.
2. **Structure-Preserving Sketching**
- **Randomized projection**: Compress gene space (~20,000 genes → 512 dimensions) using CountSketch with **leverage-score importance sampling**
- **Johnson-Lindenstrauss guarantee**: Preserves Euclidean distances between cell type signatures with high probability
- **Key innovation**: Leverage-weighted sampling amplifies rare cell type markers relative to housekeeping genes, preventing signal loss during hash collisions
3. **Spatial Graph Regularization**
- **Sparse graph Laplacian**: Constructs k-NN spatial graph (O(N) memory vs. O(N²) for dense kernels like CARD)
- **Numba-accelerated Block Coordinate Descent (BCD)**: Fast closed-form updates with non-negativity constraints
- **Linear scalability**: Spatial term complexity O(N·k) enables million-spot analysis
### Why This Works
- **Log-CPM** bounds dynamic range while preserving sparsity (log1p(0) = 0)
- **Leverage scores** decouple biological identity from population abundance—markers of rare cell types (0.1% frequency) receive equal weight to abundant types (30% frequency)
- **Sparse graph Laplacian** encodes spatial autocorrelation as a Gaussian Markov Random Field (GMRF) without dense matrix operations
---
## Benchmarks
FlashDeconv exhibits **linear O(N) scaling** for both time and memory:
| Dataset Size | Runtime | Memory | Hardware |
|:-------------|:--------|:-------|:---------|
| 10K spots | < 1 sec | < 1 GB | MacBook Pro M2 Max |
| 100K spots | ~4 sec | ~2 GB | (32GB unified memory) |
| 1M spots | ~3 min | ~21 GB | No GPU required |
**Accuracy on Synthetic Benchmarks (Spotless suite)**:
- **Pearson correlation**: 0.944 (mean across 56 datasets spanning 6 tissues)
- **RMSE**: 0.065 (median)
- **Rare cell detection (AUPR)**: 0.960 ± 0.036 (standard deviation)
**Real-world validation**:
- Mouse liver (Visium): JSD = 0.056, ranking 3rd among 13 methods
- Melanoma tumor (Visium): JSD = 0.027, ranking 5th among 13 methods
- Reference stability: Ranked 1st for robustness to different scRNA-seq protocols
FlashDeconv matches top-tier Bayesian methods (Cell2Location, RCTD) on accuracy while accelerating inference by **orders of magnitude**.
---
## API Reference
### fd.tl.deconvolve
```python
fd.tl.deconvolve(
adata_st, # Spatial AnnData
adata_ref, # Reference AnnData
cell_type_key="cell_type", # Column in adata_ref.obs
sketch_dim=512,
lambda_spatial=5000.0,
key_added="flashdeconv", # Key for results in adata_st
random_state=0, # Random seed for reproducibility
copy=False, # If True, return copy instead of inplace
)
```
**Results stored in `adata_st`:**
- `.obsm["flashdeconv"]` — Cell type proportions (DataFrame)
- `.obs["flashdeconv_dominant"]` — Dominant cell type per spot
- `.uns["flashdeconv_params"]` — Parameters used
### FlashDeconv Class
```python
class FlashDeconv:
def __init__(
self,
sketch_dim=512, # Sketch space dimension
lambda_spatial=5000.0, # Spatial regularization (or "auto")
rho_sparsity=0.01, # L1 sparsity penalty
n_hvg=2000, # Number of highly variable genes
n_markers_per_type=50, # Marker genes per cell type
spatial_method="knn", # "knn", "radius", or "grid"
k_neighbors=6, # k for k-NN graph
max_iter=100, # BCD max iterations
tol=1e-4, # Convergence tolerance
preprocess="log_cpm", # "log_cpm", "pearson", or "raw"
random_state=0, # Random seed for reproducibility
verbose=False,
): ...
def fit(self, Y, X, coords, gene_names=None, cell_type_names=None) -> self
def fit_transform(self, Y, X, coords, **kwargs) -> np.ndarray
def get_cell_type_proportions(self) -> np.ndarray
def get_abundances(self) -> np.ndarray
def get_dominant_cell_type(self) -> np.ndarray
def summary(self) -> dict
```
### Parameters
| Parameter | Type | Default | Description |
|:----------|:-----|:--------|:------------|
| `sketch_dim` | int | 512 | Dimension of sketch space (higher = more info, slower) |
| `lambda_spatial` | float or "auto" | 5000.0 | Spatial regularization strength (see Best Practices) |
| `rho_sparsity` | float | 0.01 | L1 sparsity penalty |
| `n_hvg` | int | 2000 | Number of highly variable genes to select |
| `n_markers_per_type` | int | 50 | Top markers per cell type |
| `k_neighbors` | int | 6 | Neighbors for spatial graph |
| `max_iter` | int | 100 | Maximum BCD iterations |
| `tol` | float | 1e-4 | Convergence tolerance |
| `preprocess` | str | "log_cpm" | Preprocessing: "log_cpm" (recommended), "pearson", or "raw" |
| `random_state` | int | 0 | Random seed for reproducibility (scanpy convention) |
### Attributes (After Fitting)
| Attribute | Shape | Description |
|:----------|:------|:------------|
| `beta_` | (n_spots, n_cell_types) | Raw cell type abundances |
| `proportions_` | (n_spots, n_cell_types) | Normalized proportions (sum to 1) |
| `gene_idx_` | (n_selected,) | Indices of genes used |
| `lambda_used_` | float | Actual λ value used |
| `info_` | dict | Optimization info (converged, n_iterations, final_objective) |
| `cell_type_names_` | array | Cell type names (if provided) |
---
## Input Data Formats
FlashDeconv accepts multiple input formats:
### Spatial Data (Y)
- **NumPy array**: Dense (n_spots, n_genes)
- **SciPy sparse matrix**: CSR/CSC format (recommended for Visium HD to reduce memory usage)
- **AnnData**: `.X` or specified layer (e.g., `adata.layers["counts"]`)
### Reference (X)
- **NumPy array**: Dense (n_cell_types, n_genes) signature matrix
- **AnnData**: Automatically aggregated from single-cell data via `prepare_data()` using mean expression per cell type
### Coordinates
- **NumPy array**: (n_spots, 2) for 2D spatial coordinates, or (n_spots, 3) for 3D (e.g., z-stacked sections)
- **From AnnData**: Automatically extracted from `.obsm["spatial"]`, `.obsm["X_spatial"]`, or `.obs[["x", "y"]]`
---
## Citation
If you use FlashDeconv in your research, please cite:
**Plain text:**
> Yang, C., Chen, J. & Zhang, X. FlashDeconv enables atlas-scale, multi-resolution spatial deconvolution via structure-preserving sketching. *bioRxiv* (2025). https://doi.org/10.64898/2025.12.22.696108
**BibTeX:**
```bibtex
@article{yang2025flashdeconv,
title={FlashDeconv enables atlas-scale, multi-resolution spatial deconvolution via structure-preserving sketching},
author={Yang, Chen and Chen, Jun and Zhang, Xianyang},
journal={bioRxiv},
year={2025},
doi={10.64898/2025.12.22.696108},
url={https://doi.org/10.64898/2025.12.22.696108}
}
```
---
## Contributing
Contributions are welcome! Please see [CONTRIBUTING.md](CONTRIBUTING.md) for guidelines.
---
## License
This project is licensed under the [BSD-3-Clause License](LICENSE).
---
## Related Resources
- **Paper Reproducibility:** [flashdeconv-reproducibility](https://github.com/cafferychen777/flashdeconv-reproducibility) — Complete code to reproduce all figures and benchmarks
- **Documentation:** [ReadTheDocs](https://flashdeconv.readthedocs.io) *(coming soon)*
- **Issues & Support:** [GitHub Issues](https://github.com/cafferychen777/flashdeconv/issues)
---
## Acknowledgments
We thank the developers of [Spotless](https://github.com/OmicsML/Spotless-Benchmark), [Cell2Location](https://github.com/BayraktarLab/cell2location), and [RCTD](https://github.com/dmcable/spacexr) for their benchmarking frameworks and methodological contributions to the spatial transcriptomics field.