https://github.com/elkins/synth-pdb
Generate realistic PDB files with mixed secondary structures for testing, education and bioinformatics tool development
https://github.com/elkins/synth-pdb
amino-acid-sequence bioinformatics biophysics computational-structural-biology molecular-modeling nmr-spectroscopy nmr-tools peptide peptide-sequences protein protein-data-bank protein-structure ramachandran science-education scientific-computing secondary-structure simulation structural-bioinformatics structural-biology
Last synced: 30 days ago
JSON representation
Generate realistic PDB files with mixed secondary structures for testing, education and bioinformatics tool development
- Host: GitHub
- URL: https://github.com/elkins/synth-pdb
- Owner: elkins
- License: mit
- Created: 2026-01-17T23:37:38.000Z (6 months ago)
- Default Branch: master
- Last Pushed: 2026-05-27T01:22:45.000Z (about 1 month ago)
- Last Synced: 2026-05-27T03:13:55.735Z (about 1 month ago)
- Topics: amino-acid-sequence, bioinformatics, biophysics, computational-structural-biology, molecular-modeling, nmr-spectroscopy, nmr-tools, peptide, peptide-sequences, protein, protein-data-bank, protein-structure, ramachandran, science-education, scientific-computing, secondary-structure, simulation, structural-bioinformatics, structural-biology
- Language: Python
- Homepage: https://elkins.github.io/synth-pdb/
- Size: 259 MB
- Stars: 1
- Watchers: 0
- Forks: 0
- Open Issues: 0
-
Metadata Files:
- Readme: README.md
- Changelog: CHANGELOG.md
- Contributing: CONTRIBUTING.md
- License: LICENSE
- Code of conduct: CODE_OF_CONDUCT.md
- Citation: CITATION.cff
- Security: SECURITY.md
- Zenodo: .zenodo.json
Awesome Lists containing this project
README
# synth-pdb
A command-line tool to generate Protein Data Bank (PDB) files with full atomic representation for testing, benchmarking and educational purposes.
[](https://pypi.org/project/synth-pdb/)
[](https://www.python.org/downloads/)
[](https://opensource.org/licenses/MIT)
[](https://doi.org/10.5281/zenodo.18357242)
[](https://github.com/elkins/synth-pdb/actions/workflows/test.yml)
[](https://codecov.io/gh/elkins/synth-pdb)
[](https://elkins.github.io/synth-pdb/)
[](https://github.com/astral-sh/ruff)
[](https://mypy-lang.org/)
📚 **[Read the full documentation](https://elkins.github.io/synth-pdb/)** | [Getting Started](https://elkins.github.io/synth-pdb/getting-started/quickstart/) | [API Reference](https://elkins.github.io/synth-pdb/api/overview/) | [Tutorials](examples/interactive_tutorials/gfp_molecular_forge.ipynb)
## 📚 Interactive Tutorials
### Prerequisites
- **Python 3.10+** and basic Python knowledge
- **Google Colab** account (free) or local Jupyter environment
- Specific tutorials may require domain knowledge (noted in difficulty levels)
### Tutorial Catalog
| Tutorial | Difficulty | Time | Action |
| :--- | :---: | :---: | :--- |
| [**🔬 Cryo-EM & SAXS Lab**](examples/interactive_tutorials/cryo_em_saxs_lab.ipynb) | ⭐ Beginner | 20 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/cryo_em_saxs_lab.ipynb) |
| [**🧪 The Virtual CD Lab**](examples/interactive_tutorials/virtual_cd_lab.ipynb) | ⭐ Beginner | 15 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/virtual_cd_lab.ipynb) |
| [**🤖 AI Protein Data Factory**](examples/ml_integration/ml_handover_demo.ipynb) | ⭐ Beginner | 15 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/ml_handover_demo.ipynb) |
| [**🏭 Bulk Dataset Factory**](examples/ml_integration/dataset_factory.ipynb) | ⭐ Beginner | 15 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/dataset_factory.ipynb) |
| [**🔗 Framework Handover**](examples/ml_loading/) | ⭐ Beginner | 10 min | [View JAX/PyTorch/MLX Examples](https://github.com/elkins/synth-pdb/tree/master/examples/ml_loading) |
| [**🧪 BMRB Validation Pipeline**](examples/interactive_tutorials/bmrb_validation.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/bmrb_validation.ipynb) |
| [**⭕ Macrocycle Design Lab**](examples/ml_integration/macrocycle_lab.ipynb) | ⭐⭐ Intermediate | 20 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/macrocycle_lab.ipynb) |
| [**🪞 The Mirror World Lab**](examples/interactive_tutorials/mirror_world_lab.ipynb) | ⭐⭐ Intermediate | 20 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/mirror_world_lab.ipynb) |
| [**💊 Bio-Active Hormone Lab**](examples/ml_integration/hormone_lab.ipynb) | ⭐⭐ Intermediate | 20 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/hormone_lab.ipynb) |
| [**🔍 Protein Quality Assessment**](examples/interactive_tutorials/protein_quality_assessment.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/protein_quality_assessment.ipynb) |
| [**🧠 GNN pLDDT Explorer**](examples/interactive_tutorials/gnn_plddt_explorer.ipynb) | ⭐⭐ Intermediate | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/gnn_plddt_explorer.ipynb) |
| [**🔬 The Virtual NMR Spectrometer**](examples/interactive_tutorials/virtual_nmr_spectrometer.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/virtual_nmr_spectrometer.ipynb) |
| [**🧲 RDC Alignment Tensor Explorer**](examples/interactive_tutorials/rdc_alignment_explorer.ipynb) | ⭐⭐ Intermediate | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/rdc_alignment_explorer.ipynb) |
| [**📊 RPF Score Validation**](examples/interactive_tutorials/nmr_validation_rpf.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/nmr_validation_rpf.ipynb) |
| [**🛢️ The Oil Drop Model: Hydrophobic Burial**](examples/interactive_tutorials/sasa_hydrophobic_burial.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/sasa_hydrophobic_burial.ipynb) |
| [**📡 Neural NMR Pipeline**](examples/ml_integration/neural_nmr_pipeline.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/neural_nmr_pipeline.ipynb) |
| [**🔗 The NeRF Geometry Lab**](examples/interactive_tutorials/nerf_geometry_lab.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/nerf_geometry_lab.ipynb) |
| [**📦 Modern Formats Lab**](examples/interactive_tutorials/modern_formats_lab.ipynb) | ⭐⭐ Intermediate | 15 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/modern_formats_lab.ipynb) |
| [**📏 Geometry Tools Lab**](examples/interactive_tutorials/geometry_tools_reference.ipynb) | ⭐⭐ Intermediate | 20 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/geometry_tools_reference.ipynb) |
| [**🧪 The GFP Molecular Forge**](examples/interactive_tutorials/gfp_molecular_forge.ipynb) | ⭐⭐ Intermediate | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/gfp_molecular_forge.ipynb) |
| [**⚙️ The Molecular Machine Lab**](examples/interactive_tutorials/molecular_machine_lab.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/molecular_machine_lab.ipynb) |
| [**🧠 The Prion Chameleon Lab**](examples/interactive_tutorials/prion_chameleon_lab.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/prion_chameleon_lab.ipynb) |
| [**🕸️ The NOE Network Explorer**](examples/interactive_tutorials/noe_network_explorer.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/noe_network_explorer.ipynb) |
| [**📡 NMR Relaxation Fingerprint**](examples/interactive_tutorials/nmr_relaxation_fingerprint.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/nmr_relaxation_fingerprint.ipynb) |
| [**🔭 The SAXS Shape Decoder**](examples/interactive_tutorials/saxs_shape_decoder.ipynb) | ⭐⭐ Intermediate | 25 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/saxs_shape_decoder.ipynb) |
| [**🔬 The HS-AFM Lab**](examples/interactive_tutorials/hs_afm_lab.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/hs_afm_lab.ipynb) |
| [**🎭 Protein Dynamics Theater**](examples/interactive_tutorials/protein_dynamics_theater.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/protein_dynamics_theater.ipynb) |
| [**🧬 PLM Embeddings (ESM-2)**](examples/ml_integration/plm_embeddings.ipynb) | ⭐⭐ Intermediate | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/plm_embeddings.ipynb) |
| [**📊 Ubiquitin Validation Suite**](examples/interactive_tutorials/ubiquitin_chemical_shift_validation.ipynb) | ⭐⭐⭐ Advanced | 45 min | [CS](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/ubiquitin_chemical_shift_validation.ipynb) / [J-Coupling](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/ubiquitin_j_coupling_validation.ipynb) / [RDC](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/ubiquitin_rdc_validation.ipynb) |
| [**📐 6D Orientogram Lab**](examples/ml_integration/orientogram_lab.ipynb) | ⭐⭐⭐ Advanced | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/orientogram_lab.ipynb) |
| [**🎯 The Hard Decoy Challenge**](examples/ml_integration/hard_decoy_challenge.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/hard_decoy_challenge.ipynb) |
| [**🔬 Structure Defensibility Dashboard**](examples/interactive_tutorials/structure_defensibility_dashboard.ipynb) | ⭐⭐⭐ Advanced | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/structure_defensibility_dashboard.ipynb) |
| [**🧬 Co-evolution Factory**](examples/interactive_tutorials/coevolution_msa_factory.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/coevolution_msa_factory.ipynb) |
| [**🗺️ Contact Map Fingerprinting**](examples/ml_integration/contact_map_fingerprinting.ipynb) | ⭐⭐⭐ Advanced | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/contact_map_fingerprinting.ipynb) |
| [**🧬 Co-evolutionary Fitness Landscape**](examples/ml_integration/fitness_landscape_explorer.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/fitness_landscape_explorer.ipynb) |
| [**💊 Drug Discovery Pipeline**](examples/ml_integration/drug_discovery_pipeline.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/drug_discovery_pipeline.ipynb) |
| [**🌌 AI Latent Space Explorer**](examples/interactive_tutorials/latent_space_explorer.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/latent_space_explorer.ipynb) |
| [**🏔️ The Live Folding Landscape**](examples/interactive_tutorials/folding_landscape.ipynb) | ⭐⭐⭐ Advanced | 40 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/folding_landscape.ipynb) |
| [**☁️ IDP Conformational Ensembles**](examples/interactive_tutorials/idp_ensemble_validation.ipynb) | ⭐⭐⭐ Advanced | 30 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/idp_ensemble_validation.ipynb) |
| [**🤖 AlphaFold pLDDT vs NMR S²**](examples/interactive_tutorials/alphafold_vs_nmr_dynamics.ipynb) | ⭐⭐⭐ Advanced | 35 min | [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/interactive_tutorials/alphafold_vs_nmr_dynamics.ipynb) |
### 🎓 Learning Paths
Choose a path based on your background and goals:
#### 🤖 **For ML Engineers**
*Build AI models with synthetic protein data*
1. **🤖 AI Protein Data Factory** (15 min) - Learn zero-copy data handover to PyTorch/JAX
2. **🏭 Bulk Dataset Factory** (15 min) - Generate thousands of training samples
3. **🔗 Framework Handover** (10 min) - Integrate with your ML framework
4. **🎯 Hard Decoy Challenge** (35 min) - Create negative samples for robust training
5. **🧬 PLM Embeddings (ESM-2)** (30 min) - Add evolutionary context as per-residue node features
6. **📐 6D Orientogram Lab** (30 min) - Work with rotation-invariant representations
7. **🧬 Co-evolution Factory** (35 min) - Simulate sequence evolution kernels
8. **🧠 The Prion Chameleon Lab** (25 min) - Generate high-quality misfolded decoys for robust structural scoring models
#### 🔬 **For Biophysicists**
*Understand structure, dynamics, and spectroscopy*
1. **🔗 NeRF Geometry Lab** (25 min) - Learn internal coordinate systems
2. **📏 Geometry Tools Reference** (20 min) - Kabsch, RMSD, and specialized geometry primitives
3. **🧪 Virtual CD Lab** (15 min) - Learn how secondary structure encodes Far-UV spectral signatures
4. **🔬 Virtual NMR Spectrometer** (25 min) - Predict relaxation rates and chemical shifts
5. **🧲 RDC Alignment Tensor Explorer** (30 min) - Visualize the alignment tensor and RDC physics interactively
6. **🕸️ NOE Network Explorer** (25 min) - Visualize the distance-restraint web that defines protein structure, rendered as a glowing 3D cylinder network
7. **📡 NMR Relaxation Fingerprint** (25 min) - Read protein motion from R₁/R₂/hetNOE profiles; compare 600 vs 900 MHz field dependence
8. **🔭 SAXS Shape Decoder** (25 min) - Decode protein architecture from Guinier, Kratky, and P(r) plots; distinguish folded from disordered
9. **🔬 The HS-AFM Lab** (35 min) - Generate synthetic high-speed AFM images and movies; explore tip-dilation and scanning-lag artifacts
10. **🎭 Protein Dynamics Theater** (35 min) - Compute normal modes, animate the global breathing motion, and compare NMA vs Langevin RMSF
11. **🔍 Protein Quality Assessment** (25 min) - Validate structure quality and geometry
12. **🧠 GNN pLDDT Explorer** (30 min) - Score structures with a Graph Neural Network; interpret per-residue pLDDT confidence using AlphaFold's colour scheme; compute TM-score, lDDT, and GDT-TS metrics
13. **🧪 GFP Molecular Forge** (30 min) - Explore chromophore chemistry
14. **⚙️ The Molecular Machine Lab** (25 min) - Simulate hinge motions and dynamic CD/NMR observables
15. **🧠 The Prion Chameleon Lab** (25 min) - Model alpha-to-beta transitions and infectious folding decoys
16. **🏔️ Live Folding Landscape** (40 min) - Visualize energy surfaces and Ramachandran space
17. **📡 Neural NMR Pipeline** (25 min) - Connect structure to NMR observables
18. **🧬 PLM Embeddings (ESM-2)** (30 min) - See how sequence encodes secondary structure context
19. **☁️ IDP Conformational Ensembles** (30 min) - Validate unstructured physical domains
20. **🤖 AlphaFold pLDDT vs NMR S²** (35 min) - Contrast AI rigidity with physical 15N flexibility
21. **🔬 Cryo-EM & SAXS Lab** (20 min) - Simulate 3D density maps and 1D scattering
22. **🧪 BMRB Validation Pipeline** (25 min) - Programmatic NMR validation
#### 💊 **For Drug Designers**
*Design and optimize therapeutic peptides*
1. **💊 Drug Discovery Pipeline** (35 min) - End-to-end peptide library to lead selection
2. **⭕ Macrocycle Design Lab** (20 min) - Create head-to-tail cyclic peptides
3. **💊 Bio-Active Hormone Lab** (20 min) - Model bioactive peptide hormones
4. **🪞 The Mirror World Lab** (20 min) - Design protease-resistant D-amino acid peptides
5. **🎯 Hard Decoy Challenge** (35 min) - Generate decoys for docking validation
6. **🌌 AI Latent Space Explorer** (35 min) - Navigate chemical space with ML
7. **🔬 Virtual NMR Spectrometer** (25 min) - Predict experimental observables
8. **🔬 Cryo-EM & SAXS Lab** (20 min) - Multi-modal verification of peptide folds
## Table of Contents
- [Features](#features)
- [Installation](#installation)
- [Quick Start](#quick-start)
- [Usage](#usage)
- [Command-Line Arguments](#command-line-arguments)
- [Examples](#examples)
- [ML Integration (AI Research)](#ml-integration-ai-research)
- [Validation & Refinement](#validation--refinement)
- [Output PDB Format](#output-pdb-format)
- [Scientific Context](#scientific-context)
- [Limitations](#limitations)
- [Development](#development)
- [Glossary of Scientific Terms & Acronyms](#glossary-of-scientific-terms--acronyms)
- [License](#license)
---
## 🔬 Experimental Incubator
The [`/incubator`](./incubator/) directory is our frontier for "What If?" scenarios and advanced structural biology research. This space is dedicated to developing use cases that push `synth-pdb` beyond traditional experimental boundaries:
- **Cryo-EM "Standard Candle"**: Generating atomic-resolution density maps for software benchmarking.
- **IDP Ensemble-First Validation**: Automated pipelines for modeling Intrinsically Disordered Proteins.
- **Mapping the "Dark Proteome"**: Creating hard decoys for unverified AI-predicted structures.
- **De Novo Miniprotein Forge**: Rapid prototyping for synthetic biology designs.
Check out the [Incubator README](./incubator/README.md) for the full roadmap of these experimental explorations.
---
## Features
✨ **Structure Generation**
- Full atomic representation with backbone and side-chain heavy atoms + hydrogens
- Customizable sequence (1-letter or 3-letter amino acid codes)
- Random sequence generation with uniform or biologically plausible frequencies
- **Conformational diversity**: Generate alpha helices, beta sheets, extended chains, or random conformations
- **Prompt-to-Protein Interface**: Use natural language to describe structures via `--prompt`. Supports interactive input and piping for complex requirements.
- **Backbone-Dependent Rotamers**: Side-chain conformations are selected based on local secondary structure (Helix/Sheet) to minimize steric clashes (Dunbrack library).
- **Bulk Dataset Generation**: Generate thousands of (Structure, Sequence, Contact Map) triplets for AI training via `--mode dataset`.
- **Metal Ion Coordination**: Automatic detection and structural injection of cofactors like **Zinc (Zn2+)** with physics-aware harmonic constraints.
- **Disulfide Bonds**: Automatic detection and annotation of **SSBOND** records for Cysteine pairs.
- **Salt Bridge Stabilization**: Automatic detection of ionic interactions with harmonic restraints in OpenMM.
- **Advanced Chemical Shifts**: SPARTA-lite prediction + **Ring Current Effects** (shielding/deshielding from aromatic rings).
- **Relaxation Rates**: Lipari-Szabo Model-Free formalism with **SASA-modulated Order Parameters** ($S^2$), allowing "buried" residues to be more rigid than "exposed" ones.
- **Biophysical Realism**:
- **Backbone-Dependent Rotamers**: Chi angles depend on secondary structure.
- **Pre-Proline Bias**: Residues preceding Proline automatically adopt restricted conformations (extended/beta).
- **Cis-Proline Isomerization**: X-Pro bonds can adopt cis conformations (~5% probability).
- **Post-Translational Modifications**: Support for Phosphorylation (SEP, TPO, PTR) with valid physics parameters.
- **Cyclic Peptides (Macrocycles)**: Support for **Head-to-Tail cyclization**. Closes the peptide bond between N- and C-termini using physics-based minimization.
- **NMR Functionality**: As of v1.16.0, all NMR-related features (chemical shifts, relaxation, NOEs, J-couplings) have been refactored into the separate [`synth-nmr`](https://pypi.org/project/synth-nmr/) Python package. This allows for independent use and development of NMR tools.
- **Residual Dipolar Couplings (RDCs)**: `synth_pdb.rdc` computes backbone N–H RDCs using the Saupe-matrix formalism given an alignment tensor (`Da`, `R`). Q-factor validation is demonstrated against published ubiquitin (1D3Z) data. Interactive alignment-tensor exploration is available in the `rdc_alignment_explorer.ipynb` tutorial.
- **NMR Ensemble Analysis** (`synth_pdb.ensemble`): Comprehensive tools for evaluating NMR structure bundles:
- **`DAOPCalculator`**: Dihedral Angle Order Parameter (Hyberts et al. 1992) for quantifying backbone consistency across an ensemble; includes `find_well_defined_residues` (PDBStat S(φ)+S(ψ) ≥ 1.8 convention).
- **`EnsembleStatistics`**: Typed dataclass reporting pairwise RMSD, RMSF, medoid, well-defined residues, and overall quality (Tejero et al. 2013 thresholds).
- **MSA Co-Evolution** (`synth_pdb.msa`): Generates deep multiple sequence alignments by simulating MCMC evolution on a 3D structural Potts Model — enabling zero-shot generation of DCA/AlphaFold-ready MSAs.
- Metropolis-Hastings sampling with O(1) Δ-Energy evaluation (~500× speedup).
- "Magic Step" coupled mutations for contacting residues (20% proposal rate).
- SASA selective pressure enforcing hydrophobic core isolation.
- Electrostatic salt-bridge rewards and charge-repulsion penalties in J_ij couplings.
- **Protein Language Model Embeddings** (`synth_pdb.quality.plm`): ESM-2 per-residue and pooled embeddings for zero-shot quality scoring and downstream ML tasks. Install with `pip install synth-pdb[plm]`.
- **GNN Quality Scorer** (`synth_pdb.quality.gnn`): Graph Neural Network model for structure quality assessment where nodes represent residues and edges encode sequence proximity and spatial contacts. Install with `pip install synth-pdb[gnn]`.
🚀 **High Performance Physics**
- **Hardware Acceleration**: Automatically detects and uses **GPU acceleration** (CUDA, OpenCL/Metal) if available.
- **Apple Silicon Support**: Fully supported on M1/M2/M3/M4 chips via OpenCL driver (5x speedup over CPU).
- **Vectorized Geometry**: Construction kernels are optimized with NumPy vectorization for fast validation.
- **Tunable Minimization**: Control `tolerance` and `max_iterations` to balance speed/quality for bulk datasets.
🔬 **Validation Suite**
- Bond length validation
- Bond angle validation (**Engh & Huber Z-scores**: geometry validated against the landmark 1991 standard deviations)
- Ramachandran angle checking — upgraded to **Top2018** high-resolution dataset (~15,000 chains)
- Side-Chain Rotamer validation (Chi1/Chi2 angles checked against backbone-dependent Dunbrack library)
- Steric clash detection (minimum distance + van der Waals overlap)
- Peptide plane planarity (omega angle)
- Sequence improbability detection (charge clusters, hydrophobic stretches, etc.)
- **SASA-based Burial Validation**: Shrake-Rupley algorithm (via biotite) confirming hydrophobic core formation (Kauzmann 1959)
- **`get_quality_report()`**: Multi-layered structural plausibility report covering Geometry, Physics, and Biophysics layers with peer-reviewed thresholds
⚙️ **Quality Control**
- `--best-of-N`: Generate multiple structures and select the one with fewest violations
- `--guarantee-valid`: Iteratively generate until a violation-free structure is found
- `--refine-clashes`: Iteratively adjust atoms to reduce steric clashes
- `--quality-filter`: Use Random Forest-based Structure Quality Filter to validate structure geometry
- `--quality-score-cutoff`: Set minimum confidence score for quality filter (0.0-1.0)
📝 **Reproducibility**
- Command-line parameters stored in PDB header (REMARK 3 records)
- Timestamps in generated filenames and headers
## 📚 Understanding PDB Output - Educational Guide
### Biophysical Realism
**synth-pdb** generates structures with realistic properties that mimic real experimental data:
#### 🌡️ B-factors (Temperature Factors)
**What**: Measure atomic mobility/flexibility (columns 61-66)
**Formula**: B = 8π²⟨u²⟩ (mean square displacement)
**Range**: 5-60 Ų
**Pattern**: Backbone (15-25) < Side chains (20-35) < Termini (30-50)
#### 📊 Occupancy Values
**What**: Fraction of molecules with atom at position (columns 55-60)
**Range**: 0.85-1.00
**Correlation**: High B-factor ↔ Low occupancy
**Pattern**: Backbone (0.95-1.00) > Side chains (0.85-0.95)
#### 🔄 Backbone-Dependent Rotamer Libraries
**Definition**: A **Rotamer** (Rotational Isomer) is a low-energy, stable conformation of an amino acid side chain defined by specific values of its side-chain dihedral angles ($\chi_1, \chi_2...$). Side chains are not flopping randomly; they snap into these discrete "preset" shapes.
**The "Backbone-Dependent" Twist**:
The preferred shape of a side chain strongly depends on the shape of the backbone behind it (Alpha Helix vs Beta Sheet).
* **Helix ($\alpha$)**: Side chains pack tightly. Bulky rotamers (like 'trans' chi1 for Val/Ile) often crash into the backbone (steric clash).
* **Sheet ($\beta$)**: The backbone is extended, creating more room for different rotamers.
**Implementation**: Synth-PDB uses a simplified version of the **Dunbrack Library**. It intelligently checks the backbone geometry ($\phi, \psi$) before picking a side chain shape, ensuring biophysical realism.
#### ⭕ Macrocyclization (Cyclic Peptides)
**What**: Creating a covalent bond between the N-terminal Amine and the C-terminal Carboxyl group to form a closed ring.
**Biophysical Magnitude**:
* **Conformational Entropy**: Rigidifies the peptide. A linear peptide is a "floppy" string; a cyclic peptide is a "locked" ring. This reduces the entropy loss upon binding to a receptor, significantly increasing affinity.
* **Metabolic Stability**: Most degradation in the blood happens via *exopeptidases* (enzymes that clip ends). With no ends to clip, macrocycles are much more stable and long-lived in biological systems.
* **Pre-organization**: Cyclic peptides are "pre-organized" for their biological function, making them excellent drug scaffolds.
**Coverage**: Supports **All 20 Standard Amino Acids** (including charged/polar residues).
#### 🧬 D-Amino Acids (Inverted Stereochemistry)
**What**: Mirror-images of standard L-amino acids.
**Biophysical Magnitude**:
* **Protease Resistance**: Most enzymes that degrade proteins (proteases) are "evolutionarily locked" to only recognize L-amino acids. By replacing a single L-amino acid with a D-amino acid, a peptide can become hundreds of times more stable in human blood.
* **Bacterial Cell Walls**: Bacteria uniquely use D-amino acids (like D-Ala and D-Glu) in their cross-linked peptidoglycan cell walls. This is why many antibiotics (like Penicillin) target these non-L structures.
* **Non-Natural Foldamers**: D-amino acids allow for the creation of "mirror-image" helices and unique turns (e.g., Beta-turns involving D-Pro) that are impossible with standard biology.
**Implementation**: **synth-pdb** mirrors sidechain coordinates across the N-CA-C backbone plane and uses standard PDB 3-letter codes (e.g., `DAL`, `DPH`).
#### 🧬 Secondary Structures
**What**: Regular backbone patterns (helices, sheets)
**Control**: Per-region via `--structure` parameter
**Example**: `--structure "1-10:alpha,11-15:random,16-25:alpha"`
#### 🧪 Residue-Specific Ramachandran Validation (MolProbity-Style)
> [!TIP]
> **Realism Equals Efficiency**: By using valid backbone angles (Pre-Proline bias) and correct side-chain rotamers, `synth-pdb` structures start much closer to a physical energy minimum. Validation experiments show this reduces Energy Minimization time by **>60%** due to fewer initial steric clashes.
**What**: Realistic backbone geometry validation based on amino acid type using MolProbity/Top8000 data.
- **Glycine (GLY)**: Correctly allowed in left-handed alpha region (phi > 0).
- **Proline (PRO)**: Checks against restricted phi angles.
- **General**: All other residues are checked against standard Favored/Allowed polygons.
- **Precision**: Uses point-in-polygon algorithms for accurate classification (Favored, Allowed, Outlier).
#### 📐 NeRF Geometry (The Construction Engine)
**What**: Natural Extension Reference Frame algorithm
**Term**: Building 3D structures from "Internal Coordinates" (Z-Matrix)
**Mechanism**: Places each atom (N, CA, C, O) relative to the local coordinate system of the three previous atoms.
**Educational Value**: Teaches how math converts 1D sequences + 2D angles into 3D shapes.
#### ⛓️ Metal Coordination (Cofactors)
**What**: Structural integration of inorganic ions (e.g. Zinc).
**Motifs**: Detected via ligand clustering (Cys/His sites).
**Physics**: Applied via Harmonic Constraints in Energy Minimization.
**Importance**: Models structural stability of Zinc Fingers and enzymatic sites.
#### 🧲 Salt Bridge Stabilization
**What**: Automatic detection of ionic interactions (e.g., LYS+ and ASP-).
**Criteria**: Distance-based detection between charged side-chain atoms (cutoff 5.0 Å).
**Physics**: Stabilized via harmonic restraints during energy minimization.
**Importance**: Maintains tertiary structure integrity in synthetic protein models.
#### 🔗 Disulfide Bonds (SSBOND)
**What**: Covalent bonds between Cysteine residues
**Detection**: Automatic detection of close CYS-CYS pairs (SG-SG distance 2.0-2.2 Å)
**Output**: SSBOND records added to PDB header
**Importance**: Annotates stabilizing post-translational modifications
#### ⭕ Cyclic Peptides (Macrocyclization)
**What**: Binds the N-terminal Nitrogen to the C-terminal Carbon to form a closed ring.
**Mechanism**: Uses OpenMM's physics engine to regularize the covalent bond and minimize ring strain.
**Bio-Context**: Many potent drugs (e.g., Cyclosporine) and toxins are cyclic peptides. Cyclization increases metabolic stability and reduces conformational entropy, improving binding affinity.
### Educational Philosophy & Integrity
`synth-pdb` is built on the principle of **"Code as Textbook"**.
* **Pedagogical Comments**: Key source files (`generator.py`, `test_bfactor.py`) contain detailed block comments explaining the *why* alongside the *how* (e.g., explaining Lipari-Szabo stiffness vs. B-factor flexibility).
* **Integrity Safeguards**: We include a specialized test suite (`tests/test_docs_integrity.py`) that strictly enforces the presence of these educational notes. This ensures that future refactoring never accidentally deletes the scientific context.
* **Visual Learning**: We believe that seeing is understanding. The integrated `--visualize` tool connects biophysical theory (minimized energy, restrained dynamics) to immediate visual feedback, helping visual learners grasp complex 3D relationships.
* **Universal Patterns**: The generator is tuned to reproduce universal biophysical phenomena (like terminal fraying and backbone rigidity) rather than just random noise, making it a valid tool for teaching structural biology concepts.
## Installation
### From PyPI (Recommended)
Install the latest stable release from PyPI:
```bash
pip install synth-pdb
```
This installs the `synth-pdb` package and makes the `synth-pdb` command available system-wide.
### From Source (For Development)
Install directly from the project directory:
```bash
git clone https://github.com/elkins/synth-pdb.git
cd synth-pdb
pip install .
```
### Requirements
- Python 3.10+
- NumPy
- Biotite (for residue templates and structure manipulation)
Dependencies are automatically installed with pip.
## Quick Start
Generate a simple 10-residue peptide:
```bash
synth-pdb --length 10
```
Generate and validate a specific sequence:
```bash
synth-pdb --sequence "ACDEFGHIKLMNPQRSTVWY" --validate --output my_peptide.pdb
```
Generate with mixed secondary structures and visualize:
```bash
synth-pdb --structure "1-10:alpha,11-20:beta" --visualize
```
Generate the best of 10 attempts with clash refinement:
```bash
synth-pdb --length 20 --best-of-N 10 --refine-clashes 5 --output refined_peptide.pdb
```
## 🤖 Feature Spotlight: AI Model Support & Hard Decoys
Generating "good" structures is only half the battle. To train robust AI models (like AlphaFold-3 or RosettaFold), researchers need **High-Quality Negative Samples**—structures that look physically plausible but are biologically or topologically incorrect.
**Synth-PDB** provides three powerful mechanisms for generating these "Hard Decoys":
### 1. Sequence Threading (Fold Mismatch)
Force a specific sequence onto the backbone "fold" of a completely different sequence. This creates a realistic-looking structure where the side-chain packing is fundamentally incompatible with the backbone.
```bash
# Thread Poly-Ala sequence onto a backbone generated for Poly-Pro
synth-pdb --mode decoys --sequence AAAAA --template-sequence PPPPP --hard
```
### 2. Torsion Angle Drift (Conformational Noise)
Add controlled, random noise to ideal Ramachandran angles. This creates "near-native" decoys—structures that are *almost* correct but have subtle, realistic errors.
```bash
# Add 5 degrees of maximum drift to all phi/psi angles
synth-pdb --mode decoys --drift 5.0
```
### 3. Label Shuffling (Sequence Mismatch)
Generate a perfectly valid structure for a sequence, then randomly shuffle the identity of the residues in the final PDB. This tests if an AI model can detect that a residue (e.g., Trp) is in an environment meant for another (e.g., Gly).
```bash
synth-pdb --mode decoys --sequence ACDEF --hard --shuffle-sequence
```
---
## 🌟 Feature Spotlight: "Spectroscopically Realistic" Dynamics
Most synthetic PDB generators create static bricks. They might create reasonable geometry, but the "B-factor" column (Column 11) is often just zero or random noise.
**Synth-PDB is different.** It simulates the **physics of protein motion** to generate a unified model of structure AND dynamics.
### The "Structure-Dynamics Link"
We implement the **Lipari-Szabo Model-Free formalism** (Nobel-adjacent physics) directly into the generator:
1. **Structure Awareness**: The engine analyzes the generated geometry (`alpha-helix` vs `random-coil`).
2. **Order Parameter ($S^2$) Prediction**: It assigns specific rigidity values:
* **Helices**: $S^2 \approx 0.85$ (Rigid H-bond network)
* **Loops**: $S^2 \approx 0.65$ (Flexible nanosecond motions)
* **Termini**: $S^2 \approx 0.45$ (Disordered fraying)
3. **Unified Output**:
* **PDB B-Factors**: Calculated via $B \propto (1 - S^2)$. When you visualize the PDB in PyMOL, flexible regions *visually* appear thicker/redder, matching real crystal data distributions.
* **NMR Relaxation**: $R_1, R_2, NOE$ rates are calculated from the *same* parameters.
**Why this matters**:
> "The correlation between NMR order parameters ($S^2$) and crystallographic B-factors is a bridge between solution-state and solid-state dynamics." — *Fenwick et al., PNAS (2014)*
This feature allows you to test **bioinformatics pipelines** that rely on correlation between sequence, structure, and experimental observables, without needing expensive Molecular Dynamics (MD) simulations.
### 4. Relax (Simulate Dynamics)
Generate relaxation rates ($R_1, R_2, NOE$) with **realistic internal dynamics**:
```bash
python main.py relax --input output/my_peptide.pdb --output output/relaxation_data.nef --field 600 --tm 10.0
```
This module now implements the **Lipari-Szabo Model-Free** formalism with structure-based Order Parameter ($S^2$) prediction:
* **Helices/Sheets**: $S^2 \approx 0.85$ (Rigid, high $R_1/R_2$)
* **Loops/Turns**: $S^2 \approx 0.65$ (Flexible, lower $R_1/R_2$)
* **Termini**: $S^2 \approx 0.45$ (Highly disordered)
This creates realistic "relaxation gradients" along the sequence, perfect for testing dynamics software.
## 🚀 Quick Visual Demo
Want to see the **Physics + Visualization** capabilities in action?
Run this command to generate a **Leucine Zipper** (classic alpha helix), **minimize** its energy using OpenMM, and immediately **visualize** it in your browser:
```bash
synth-pdb --sequence "LKELEKELEKELEKELEKELEKEL" --conformation alpha --minimize --visualize
```
This effectively demonstrates:
1. **Generation**: Creating the alpha-helical backbone.
2. **Minimization**: "Relaxing" the structure (geometry regularization).
3. **Visualization**: Launching the interactive 3D viewer.
## Usage
### Command-Line Arguments
#### **Structure Definition**
- `--length `: Number of residues in the peptide chain
- Type: Integer
- Default: `10`
- Example: `--length 50`
- `--sequence `: Specify an exact amino acid sequence
- Formats:
- 1-letter codes: `"ACDEFG"`
- 3-letter codes: `"ALA-CYS-ASP-GLU-PHE-GLY"`
- Overrides `--length`
- Example: `--sequence "MVHLTPEEK"`
- `--plausible-frequencies`: Use biologically realistic amino acid frequencies for random generation
- Based on natural protein composition
- Ignored if `--sequence` is provided
- `--conformation \u003cCONFORMATION\u003e`: Secondary structure conformation to generate
- Options: `alpha`, `beta`, `ppii`, `extended`, `random`
- Default: `alpha` (alpha helix)
- Choices:
- `alpha`: Alpha helix (φ=-57°, ψ=-47°)
- `beta`: Beta sheet (φ=-135°, ψ=135°)
- `ppii`: Polyproline II helix (φ=-75°, ψ=145°)
- `extended`: Extended/stretched conformation (φ=-120°, ψ=120°)
- `random`: Random sampling from allowed Ramachandran regions
- Example: `--conformation beta`
#### 🤖 AI & Machine Learning: Bulk Dataset Generation
`synth-pdb` serves as valid data generator for training Deep Learning models (GNNs, Transformers, Diffusion Models). It can generate massive, diverse, and labeled datasets.
**Command:**
```bash
synth-pdb --mode dataset --dataset-format npz --num-samples 1000 --output my_training_data
```
**Features:**
* **Formats**:
* `npz`: (Recommended) Compressed NumPy archives. Contains `coords` (L,5,3), `sequence` (One-hot), and `contact_map` (LxL). Ideal for PyTorch/TensorFlow dataloaders.
* `pdb`: Writes individual PDB files and CASP contact maps (slower, for legacy tools).
* **Multiprocessing**: Automatically uses all available CPU cores.
* **Manifest**: Generates a `dataset_manifest.csv` tracking all samples and their metadata (split, length, conformation).
**Output Structure (`--dataset-format npz`)**:
```
my_training_data/
├── dataset_manifest.csv
├── train/
│ ├── synth_000001.npz
│ ├── synth_000002.npz
│ ...
└── test/
├── synth_000801.npz
...
```
### 🔍 Visualization & Analysis
#### **Validation & Quality Control**
- `--validate`: Run validation checks on the generated structure
- Checks: bond lengths, bond angles, Ramachandran, steric clashes, peptide planes, sequence improbabilities
- Reports violations to console
- `--guarantee-valid`: Generate structures until one with zero violations is found
- Implies `--validate`
- Use with `--max-attempts` to limit iterations
- Example: `--guarantee-valid --max-attempts 100`
- `--max-attempts `: Maximum generation attempts for `--guarantee-valid`
- Default: `100`
- `--best-of-N `: Generate N structures and select the one with fewest violations
- Implies `--validate`
- Overrides `--guarantee-valid`
- Example: `--best-of-N 20`
- `--refine-clashes `: Iteratively adjust atoms to reduce steric clashes
- Applies after structure selection
- Iterates until improvements stop or max iterations reached
- Example: `--refine-clashes 10`
#### **Structure Quality Filter (Random Forest)**
> [!NOTE]
> Despite the flag name history, this feature uses a **classical Random Forest classifier** (scikit-learn), not a neural network or generative AI. It scores structures on geometric quality metrics derived from Ramachandran angles, steric clashes, bond lengths, and radius of gyration.
- `--quality-filter`: Enable the **Structure Quality Filter** to screen generated structures.
- Using a Random Forest classifier trained on thousands of samples, this filter automatically rejects "low quality" structures (clashing, distorted geometry).
- It considers Ramachandran angles, steric clashes, bond lengths, and radius of gyration.
- Useful for filtering out failed minimization attempts in bulk generation.
- `--quality-score-cutoff `: Minimum probability score (0.0-1.0) for a structure to be considered "Good".
- Higher values = stricter filtering (fewer false positives, more false negatives).
- Default: `0.5`
- Example: `--quality-score-cutoff 0.8` (Only keep highly confident good structures)
- Scores below `0.5` are typically rejected as "Bad".
#### **Physics & Advanced Refinement **
- `--minimize`: Run physics-based energy minimization (OpenMM).
- Defaults to implicit solvent (OBC2) and AMBER forcefield.
- Highly recommended for "realistic" geometry.
- Example: `--minimize`
- `--solvent `: Specify the solvent model for minimization/equilibration.
- Options: `obc2` (default), `obc1`, `gbn`, `gbn2`, `hct`, `explicit`
- Example: `--solvent explicit` (simulates a TIP3P water box)
- `--solvent-padding `: Padding distance (in nm) for the explicit water box.
- Default: `1.0`
- Example: `--solvent-padding 1.5`
- `--keep-solvent`: Retain the generated water molecules (HOH) in the final PDB file.
- Default: False (water is stripped for cleaner outputs)
- `--optimize`: Run Monte Carlo side-chain optimization.
- Reduces steric clashes by rotating side chains.
- Example: `--optimize`
- `--forcefield `: Specify OpenMM forcefield.
- Default: `amber14-all.xml`
- Example: `--forcefield amber14-all.xml`
- Default: `amber14-all.xml`
- `--minimization-k `: Energy minimization tolerance (kJ/mole/nm).
- Higher values = Faster but less precise.
- Recommended for bulk generation: `100.0`
- Default: `10.0` (High Precision)
- `--minimization-max-iter `: Max iterations for minimization.
- `0` = Unlimited (Convergence based on tolerance)
- Recommended for bulk generation: `1000`
- Default: `0`
#### **Synthetic NMR Data**
> **📦 NMR Functionality Powered by [`synth-nmr`](https://github.com/elkins/synth-nmr)**
> As of version 1.17.0, all NMR-related functionality (NOE calculation, relaxation rates, chemical shifts, J-couplings) is provided by the standalone [`synth-nmr`](https://pypi.org/project/synth-nmr/) package. This package can be used independently for NMR data generation in your own projects. The integration is fully backward compatible—all existing code continues to work without changes.
- `--gen-nef`: Generate synthetic NOE restraints in NEF format.
- Scans structure for H-H pairs < cutoff.
- Outputs `.nef` file.
- Note: Requires hydrogens (use with `--minimize` or internal default).
- `--noe-cutoff `: Cutoff distance for NOEs in Angstroms.
- Default: `5.0`
- Example: `--noe-cutoff 6.0`
- `--nef-output `: Custom output filename for NEF.
#### **Synthetic Relaxation Data **
- `--gen-relax`: Generate synthetic NMR relaxation data ($R_1, R_2, \{^1H\}-^{15}N\ NOE$) in NEF format.
- Calculates Model-Free parameters ($S^2 \approx 0.85$ for core, $0.5$ for flexible termini).
- Outputs `_relax.nef` file.
- **Physics Note**: $NOE$ values depend on tumbling time, not just internal flexibility.
- `--field `: Proton Larmor frequency in MHz.
- Default: `600.0`
- Calculates proper spectral density frequencies for this field.
- `--tumbling-time `: Global rotational correlation time ($\tau_m$) in nanoseconds.
- Default: `10.0`
- Controls the overall magnitude of relaxation rates. Larger proteins have larger $\tau_m$.
#### **Constraints Export **
- `--export-constraints `: Export contact map constraints for modeling/folding.
- Useful for checking agreement with AlphaFold/CASP predictions.
- Outputs a file containing residue-residue contacts.
- Example: `--export-constraints constraints.casp`
- `--constraint-format {casp,csv}`: Format for the exported constraints.
- `casp`: Critical Assessment of Structure Prediction (RR) format.
- `csv`: Comma-separated values (i, j, distance).
- Default: `casp`
- `--constraint-cutoff `: Distance cutoff for defining binary contacts (Angstroms).
- Default: `8.0`
#### **Torsion Angle Export **
- `--export-torsion `: Export backbone torsion angles (Phi, Psi, Omega) for every residue.
- Useful for training ML models on backbone geometry.
- Outputs a CSV or JSON file.
- Example: `--export-torsion angles.csv`
- `--torsion-format {csv,json}`: Format for the exported data.
- Default: `csv`
#### **Synthetic MSA (Evolution) **
- `--gen-msa`: Generate a Multiple Sequence Alignment (MSA) by simulating neutral drift.
- Conserves hydrophobic core residues while mutating surface residues.
- Outputs a FASTA file useful for testing co-evolution signals in AI models.
- `--msa-depth `: Number of sequences to generate.
- Default: `100`
- `--mutation-rate `: Probability of mutation per position per sequence.
- Default: `0.1` (10% divergence per sequence).
#### **Distogram Export (Spatial Relationships) **
- `--export-distogram `: Export NxN Distance Matrix representing the protein geometry.
- Rotation-invariant representation ideal for AI model training/validation.
- Supports `json`, `csv`, or `npz` (NumPy) formats.
- Example: `--export-distogram dist.json`
- `--distogram-format {json,csv,npz}`: Output format.
- Default: `json`
#### **Biophysical Realism (Physics) **
- `--ph `: Set pH for titration (default 7.4).
- Automatically adjusts Histidine protonation (`HIS` $\rightarrow$ `HIP` if pH < 6.0).
- Critical for realistic electrostatics and NMR chemical shifts.
- `--cap-termini`: Add terminal blocking groups.
- N-terminus: Acetyl (`ACE`)
- C-terminus: N-methylamide (`NME`)
- Removes charged termini ($\text{NH}_3^+$/$\text{COO}^-$) for realistic peptide modeling.
- `--cyclic`: Generate a **Head-to-Tail cyclic peptide**.
- Connects the N-terminus and C-terminus with a covalent peptide bond.
- **Requirement**: Automatically implies `--minimize` to ensure proper closure.
- **Incompatibility**: Disables `--cap-termini`.
- `--equilibrate`: Run Molecular Dynamics (MD) equilibration.
- Simulates the protein at **300 Kelvin** (solution state).
- Uses Langevin Dynamics to shake atoms out of local minima.
- Generates a "thermalized" structure closer to NMR conditions.
- Options: `--md-steps ` (default 1000, $\approx$ 2 ps).
- `--metal-ions {auto,none}`: Control metal ion coordination.
- `auto` (default): Scans for binding sites and injects ions.
- `none`: Disables automatic coordination.
- `--phosphorylation-rate `: Probability of phosphorylating S/T/Y residues.
- Value between 0.0 and 1.0.
- Converts SER->SEP, THR->TPO, TYR->PTR.
- Mimics kinase activity for regulatory simulation.
- Example: `--phosphorylation-rate 0.5`
- `--cis-proline-frequency `: Probability of X-Pro peptide bond being Cis.
- Default: `0.05` (5%)
- Cis-Proline is critical for tight turns and folding.
- Set to `0.0` for all-Trans, `1.0` for all-Cis.
#### **Bulk Dataset Generation (AI)**
- `--mode dataset`: Enable bulk generation mode.
- `--num-samples `: Number of samples to generate (default 100).
- `--min-length `, `--max-length `: Range for random sequence lengths (default 10-50).
- `--train-ratio `: Fraction of samples for the training set (default 0.8).
- `--output `: Directory to save the dataset.
#### **Output Options**
- `--output `: Custom output filename
- If omitted, auto-generates: `random_linear_peptide__.pdb`
- Example: `--output my_protein.pdb`
- `--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}`: Logging verbosity
- Default: `INFO`
- Use `DEBUG` for detailed validation reports
- `--seed `: Random seed for reproducible generation
- Default: `None` (Random)
- Example: `--seed 42`
- Guarantees identical output for the same command.
- `--help`: Show the help message and exit.
### Examples
#### Basic Generation
```bash
# Simple 25-residue peptide
synth-pdb --length 25
# Custom sequence with validation
synth-pdb --sequence "ELVIS" --validate --output elvis.pdb
# Use biologically realistic frequencies
synth-pdb --length 100 --plausible-frequencies
# Generate a random 20-residue alpha helix
synth_pdb --length 20 --conformation alpha --output random_helix.pdb
# Generate a high-quality, physically realistic structure (Recommended)
# Includes: Minimization, Terminal Capping, and Thermal Equilibration (MD)
synth_pdb --length 20 --minimize --cap-termini --equilibrate --output best_structure.pdb
# Generate beta sheet conformation
synth-pdb --length 20 --conformation beta --output beta_sheet.pdb
# Generate extended conformation
synth-pdb --length 15 --conformation extended
# Generate random conformation (mixed alpha/beta regions)
synth-pdb --length 30 --conformation random
# 🤖 Bulk dataset generation for AI training
synth-pdb --mode dataset --num-samples 500 --min-length 10 --max-length 40 --output ./my_dataset
# ⛓️ Generate a Zinc Finger with structural cofactors
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --minimize --metal-ions auto --output zinc_finger.pdb
```
#### Quality Control
```bash
# Generate until valid (may take time!)
synth-pdb --length 15 --guarantee-valid --max-attempts 200 --output valid.pdb
# Best of 50 attempts
synth-pdb --length 20 --best-of-N 50 --output best_structure.pdb
```
#### Explicit Solvent & Hardware Testing
Simulate your protein in a realistic water box (TIP3P) for high-fidelity physics or export the explicit solvent map for downstream molecular dynamics.
```bash
# Basic explicit solvent: generate a small peptide and pad with 1.2 nm of water.
# By default, synth-pdb strips the water atoms before saving the final clean PDB.
synth-pdb --sequence ALA-PRO-GLY --minimize --solvent explicit --solvent-padding 1.2 --output small_peptide.pdb
# Retain the water box: save the entire simulated system (protein + thousands of HOH atoms)
synth-pdb --sequence TRP-TYR-PHE --minimize --solvent explicit --solvent-padding 1.5 --keep-solvent --output full_water_box.pdb
# 🚀 EXTREME Hardware Limit Test
# Generate a large 50-residue sequence, bury it in a massive 2.5 nm water box,
# and run 10,000 steps of Langevin Dynamics equilibration.
# WARNING: This will generate >50,000 atoms and heavily tax your CPU/GPU!
synth-pdb --length 50 --conformation random --minimize --equilibrate --md-steps 10000 --solvent explicit --solvent-padding 2.5 --keep-solvent --output extreme_limit_test.pdb
```
## ML Integration (AI Research)
**synth-pdb** is designed to be a high-performance "Data Factory" for Training Protein AI models. It can generate thousands of unique, physically plausible protein structures in seconds—bypassing the bottleneck of parsing millions of PDB files from disk.
### 🤖 The Batch Walk (Vectorized Performance)
Using the `BatchedGenerator` module, the tool uses SIMD/Vectorized math (NeRF algorithm) to build peptide backbones in parallel.
### ⚡ Zero-Copy Handover
Transition from biological coordinates to Deep Learning tensors instantly. Our `BatchedPeptide` output is **C-Contiguous**, allowing tools like PyTorch and JAX to map the memory without copying data.
```python
from synth_pdb.batch_generator import BatchedGenerator
import torch
# Generate 1,000 structures in milliseconds
bg = BatchedGenerator("ALA-GLY-SER-TRP", n_batch=1000)
batch = bg.generate_batch()
# Instant PyTorch Handover (Shared RAM)
coords_tensor = torch.from_numpy(batch.coords).float()
```
### 🚀 Try it in the Cloud
- **AI Protein Data Factory:** [](https://colab.research.google.com/github/elkins/synth-pdb/blob/master/examples/ml_integration/ml_handover_demo.ipynb)
### 🧩 Framework Specifics
For detailed examples of how to load generated data into your favorite framework without any performance overhead, see our specialized handover notebooks:
- [JAX Handover](examples/ml_loading/jax_handover.ipynb) - Zero-copy using `jax.numpy.asarray`.
- [PyTorch Handover](examples/ml_loading/pytorch_handover.ipynb) - Unified memory mapping with `torch.from_numpy`.
- [MLX Handover](examples/ml_loading/mlx_handover.ipynb) - Optimized for Apple Silicon (M-series CPUs/GPUs).
#### Quality Control (Continued)
```bash
# Refine steric clashes (5 iterations)
synth-pdb --length 30 --refine-clashes 5 --output refined.pdb
# Combined: best of 10 + refinement
synth-pdb --length 25 --best-of-N 10 --refine-clashes 3 --output optimized.pdb
```
#### Biologically-Inspired Examples
Generate structures that mimic real protein motifs for educational demonstrations:
```bash
# Collagen-like triple helix motif (polyproline II)
# Collagen is rich in proline and glycine with PPII conformation
synth-pdb --sequence "GPGPPGPPGPPGPPGPPGPP" --conformation ppii --output collagen_like.pdb
# Silk fibroin-like beta sheet
# Silk proteins contain repeating (GAGAGS) motifs forming beta sheets
synth-pdb --sequence "GAGAGSGAGAGSGAGAGS" --conformation beta --output silk_like.pdb
# Amyloid fibril-like beta structure
# Amyloid fibrils are rich in beta sheets, often with hydrophobic residues
synth-pdb --sequence "LVEALYLVCGERGFFYTPKA" --conformation beta --best-of-N 10 --output amyloid_like.pdb
# Leucine zipper motif (alpha helix)
# Leucine zippers are alpha-helical with leucine repeats every 7 residues
synth-pdb --sequence "LKELEKELEKELEKELEKELEKEL" --conformation alpha --output leucine_zipper.pdb
# Intrinsically disordered region (random conformation)
# IDRs lack stable structure, rich in charged/polar residues
synth-pdb --sequence "GGSEGGSEGGSEGGSEGGSE" --conformation random --output disordered_region.pdb
# Transmembrane helix-like structure (extended alpha helix)
# Membrane-spanning regions are often long alpha helices with hydrophobic residues
synth-pdb --sequence "LVIVLLVIVLLVIVLLVIVL" --conformation alpha --output transmembrane_like.pdb
# Beta-turn rich structure (mixed conformations)
# Proline and glycine favor turns and loops
synth-pdb --sequence "GPGPGPGPGPGPGPGP" --conformation random --output beta_turn_rich.pdb
# Elastin-like peptide (extended/random)
# Elastin contains repeating VPGVG motifs with flexible structure
synth-pdb --sequence "VPGVGVPGVGVPGVGVPGVG" --conformation extended --output elastin_like.pdb
# Antimicrobial peptide-like (alpha helix)
# Many AMPs are short amphipathic alpha helices
synth-pdb --sequence "KWKLFKKIGAVLKVL" --conformation alpha --validate --output amp_like.pdb
# Zinc finger motif-like (mixed structure)
# Zinc fingers have beta sheets and alpha helices
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --conformation random --best-of-N 5 --output zinc_finger_like.pdb
```
**Educational Notes:**
- These examples demonstrate **sequence-structure relationships**
- Real proteins would have more complex tertiary structures and post-translational modifications
- Use these for teaching secondary structure concepts, not for actual molecular modeling
- Combine with `--validate` to show how different conformations affect structural quality
- Try `--best-of-N` and `--refine-clashes` to explore quality control strategies
#### Visualization-Optimized Examples
These examples are specifically designed to look great in the 3D viewer with `--visualize`:
```bash
# 🧬 Compact Alpha Helix (BEST for visualization)
# Short, tight helix - perfect for interactive viewing
synth-pdb --length 15 --conformation alpha --visualize
# 🔗 Helix-Turn-Helix DNA-Binding Motif
# Classic protein architecture with two helices and a turn
synth-pdb --sequence "AAAAAAGGGAAAAA" --structure "1-6:alpha,7-9:random,10-14:alpha" --visualize
# 🧬 "Textbook" Stabilized Alpha Helix (Salt Bridges)
# Demonstrates charge pairs (Glu-Lys) stabilizing the backbone (i, i+4)
# Use --minimize to geometry-optimize these ionic interactions
synth-pdb --sequence "EAAKEAAKEAAKEAAK" --conformation alpha --minimize --cap-termini --visualize
# 🔗 Zinc Finger with Metal Coordination
# See the Zinc ion (Zn2+) automatically coordinated by Cys/His residues!
# The --minimize flag applies harmonic constraints to the metal center.
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --structure "1-10:beta,11-20:alpha" --metal-ions auto --minimize --visualize
# 🎀 Refined Beta Hairpin
# Two antiparallel beta strands connected by a turn, relaxed with physics
synth-pdb --sequence "VVVVVGGVVVVV" --structure "1-5:beta,6-8:random,9-12:beta" --minimize --visualize
# 🧪 Polyproline II Helix (Collagen-like)
# Left-handed helix, compact and visually distinct
synth-pdb --sequence "GPGPPGPPGPPGPP" --conformation ppii --minimize --visualize
# 🧪 The "Kitchen Sink" (Features Demo)
# Combines distinct secondary structures (Helix, Sheet) with a Type I Beta Turn and PTMs.
# Look for the magenta helix, purple turn, and orange phosphorylated residues (SEP/TPO/PTR).
synth-pdb --length 25 --structure "1-10:alpha,11-14:typeI,15-25:beta" --phosphorylation-rate 0.3 --visualize
# ⭕ The "Molecular Hoop" (Macrocycle)
# A simple flexible ring of Glycines. Perfect for visualizing ring closure.
synth-pdb --sequence "GGGGGGGGGGGG" --cyclic --minimize --visualize
```
**Visualization Tips:**
- **Best conformations for viewing**: `alpha` (most compact), `ppii` (distinctive shape)
- **Optimal length**: 10-20 residues for clear visualization
- **In the viewer**: Use "Cartoon" style and "Spectrum" color for best results
- **Interactive**: Rotate with left-click, zoom with scroll, pan with right-click
#### Mixed Secondary Structures
The `--structure` parameter enables creation of realistic protein-like structures with different conformations in different regions:
```bash
# Helix-turn-helix DNA-binding motif
# Two alpha helices connected by a flexible turn region, minimized for realism
synth-pdb --length 25 --structure "1-10:alpha,11-15:random,16-25:alpha" --minimize --output helix_turn_helix.pdb
# Beta-alpha-beta fold unit
# Common protein architecture with sheet-helix-sheet
synth-pdb --length 30 --structure "1-10:beta,11-15:random,16-25:alpha,26-30:beta" --minimize --output bab_fold.pdb
# Zinc finger with realistic structure
# Beta sheet + alpha helix (actual zinc finger architecture)
synth-pdb --sequence "CPHCGKSFSQKSDLVKHQRT" --structure "1-5:beta,6-10:random,11-20:alpha" --minimize --output zinc_finger_realistic.pdb
# Immunoglobulin domain
# Multiple beta sheets connected by loops (antibody-like)
synth-pdb --length 40 --structure "1-8:beta,9-12:random,13-20:beta,21-24:random,25-32:beta,33-40:random" --minimize --output ig_domain.pdb
# Coiled-coil with flexible linker
# Two helical regions connected by disordered linker
synth-pdb --length 50 --structure "1-20:alpha,21-30:random,31-50:alpha" --minimize --output coiled_coil.pdb
# Intrinsically disordered region with structured domain
# Disordered N-terminus, structured C-terminus (common in signaling proteins)
synth-pdb --length 40 --structure "1-15:random,16-40:alpha" --minimize --output idr_with_domain.pdb
# Collagen-like with flexibility
# PPII helix with occasional flexible regions (more realistic than uniform)
synth-pdb --sequence "GPGPPGPPGPPGPPGPPGPP" --structure "1-6:ppii,7-9:random,10-20:ppii" --output collagen_flexible.pdb
# Beta-hairpin motif
# Two antiparallel beta strands connected by a turn
synth-pdb --length 20 --structure "1-7:beta,8-12:random,13-20:beta" --refine-clashes 5 --output beta_hairpin.pdb
```
**Why This Matters:**
- Real proteins have **mixed secondary structures**, not uniform conformations
- These examples are much more realistic than single-conformation structures
- Useful for teaching protein architecture and domain organization
- Great for testing structure analysis tools with realistic inputs
- Demonstrates how sequence and structure work together
#### Detailed Educational Case Studies
These comprehensive examples demonstrate how to use `synth-pdb` to model specific biological features found in well-known proteins.
**1. Glucagon (Alpha Helix Hormone)**
*29 residues | PDB: 1GCN*
Glucagon is a peptide hormone that raises glucose levels. It folds into a characteristic alpha helix.
```bash
synth-pdb --sequence HSQGTFTSDYSKYLDSRRAQDFVQWLMNT --conformation alpha --refine-clashes 0 --output glucagon.pdb
```
*Educational Concept*: Studying alpha-helical packing and amphipathicity.
**2. Melittin (Bent Helix / Hinge)**
*26 residues | PDB: 2MLT*
The principal toxin in bee venom. It forms two alpha helices separated by a "hinge" region, allowing it to puncture membranes.
```bash
synth-pdb --sequence GIGAVLKVLTTGLPALISWIKRKRQQ --structure "1-11:alpha,12-14:random,15-26:alpha" --refine-clashes 50 --output melittin.pdb
```
*Educational Concept*: Modeling non-linear secondary structures and flexible linkers (hinges).
**3. Bovine Pancreatic Trypsin Inhibitor (BPTI) (Disulfide Bonds)**
*58 residues | PDB: 1BPI*
A classic model for protein folding studies ("The Hydrogen Atom of Protein Folding"). It is stabilized by three disulfide bonds.
```bash
synth-pdb --sequence RPDFCLEPPYTGPCKARIIRYFYNAKAGLCQTFVYGGCRAKRNNFKSAEDCMRTCGGA --conformation random --minimize --visualize --output bpti.pdb
```
*Educational Concept*: Automatic detection of disulfide bonds (`SSBOND` records). The `--minimize` flag brings cysteine sulfurs into proper bonding distance (2.0 Å).
**4. Ubiquitin (Complex Mixed Fold)**
*76 residues | PDB: 1UBQ*
A highly conserved regulatory protein with a complex mixed alpha/beta fold (beta grasp fold).
```bash
synth-pdb --sequence MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG --structure "1-7:beta,12-16:beta,23-34:alpha,41-45:beta,48-49:beta,56-59:alpha,66-70:beta" --minimize --best-of-N 5 --output ubiquitin.pdb
```
*Educational Concept*: Generating complex, multi-domain topologies. Physics-based minimization (`--minimize`) resolves steric clashes better than geometric heuristics alone.
**5. SFTI-1 (Sunflower Trypsin Inhibitor)**
*14 residues | PDB: 1SFI*
A small, potent protease inhibitor that is both **cyclic** and stabilized by a **disulfide bond**.
```bash
synth-pdb --sequence "GRCTKSIPPICFPD" --cyclic --minimize --visualize --output sfti1.pdb
```
*Educational Concept*: Combining multiple stabilizing modifications (**Cyclization** + **Disulfide Bonds**) to create a rigid, functional scaffold.
**6. Gramicidin S (D-Amino Acid Antibiotic)**
*10 residues | PDB: 1TK2*
A powerful cyclic antibiotic produced by soil bacteria. It contains the rare **D-Phenylalanine** (`D-PHE`) which is critical for its "beta-sheet-like" hairpins.
```bash
synth-pdb --sequence "VAL-ORN-LEU-D-PHE-PRO-VAL-ORN-LEU-D-PHE-PRO" --cyclic --minimize --visualize --output gramicidin_s.pdb
```
*Note: This utilizes ORN (Ornithine) if supported, or sub for LYS. The key is the D-PHE residue.*
*Educational Concept*: Using D-amino acids to induce specific turns and achieve antimicrobial activity through membrane disruption.
#### 🏗️ "Architectural" Protein Examples (The Giants)
These larger structures demonstrate domain organization and fibrous protein architectures.
**1. "Synthetic Spectrin" (Multi-Domain Repeat)**
*~150 Residues*
Spectrin is a cytoskeletal protein made of repeating triple-helical bundles. We can simulate a simplified version: three distinct alpha-helical domains connected by flexible linkers.
```bash
synth-pdb --length 150 --structure "1-40:alpha,41-50:random,51-90:alpha,91-100:random,101-140:alpha,141-150:random" --minimize --visualize --output synthetic_spectrin.pdb
```
*Educational Concept*: Demonstrates "beads on a string" domain organization and stable inter-domain flexibility.
**2. "Titin Segment" (Poly-Beta Repeat)**
*~120 Residues*
Titin acts as a molecular spring in muscle, made of distinct Ig-like (beta sheet) domains.
```bash
synth-pdb --length 120 --structure "1-30:beta,31-40:random,41-70:beta,71-80:random,81-110:beta,111-120:random" --minimize --visualize --output titin_segment.pdb
```
*Educational Concept*: Shows distinct rigid beta-regions separated by disordered "hinges", mimicking force-bearing structural proteins.
**3. "Giant Coiled-Coil" (The Molecular Rod)**
*~100 Residues*
A super-long continuous alpha helix, modeled after Myosin tails or Tropomyosin.
```bash
synth-pdb --sequence "LKELEKELEKELEKELEKELEKELEKELEKELEKELEKELEKELEKELEKE" --conformation alpha --minimize --visualize --output long_coil.pdb
```
*Educational Concept*: A massive, rigid rod where the helical groove is clearly visible. Excellent for demonstrating persistence length.
**4. "Synthetic Antibody" (The Ultimate Stress Test)**
*450 Residues*
Empirical simulation of a full IgG Heavy Chain: 4 Beta-sandwich domains (VH, CH1, CH2, CH3) connected by linkers.
```bash
synth-pdb --length 450 --structure "1-100:beta,101-110:random,111-210:beta,211-230:random,231-330:beta,331-340:random,341-440:beta,441-450:random" --minimize --visualize --output synthetic_antibody.pdb
```
*Note*: This is a computationally intensive task! Energy minimization for ~7000 atoms may take several minutes.
*Educational Concept*: Simulating multi-domain packing and the flexibility of the hinge region (residues 211-230).
#### For Structural Biologists
```bash
# All natural amino acids with validation report
synth-pdb --sequence "ACDEFGHIKLMNPQRSTVWY" --validate --log-level DEBUG
# Test structure for MD simulation pipeline
synth-pdb --length 50 --guarantee-valid --max-attempts 500 --output test_md.pdb
# Benchmark structure with known violations (good for testing validators)
synth-pdb --length 100 --validate --output benchmark.pdb
```
#### The "Power User" Pipeline ⚡️
Combine all features to simulate a complete NMR structure determination workflow:
1. **Generate** a sequence.
2. **Fold** it (alpha helix).
3. **Refine** geometry (minimization).
4. **Simulate** experimental data (NOEs and Relaxation).
5. **Visualize** the result.
```bash
synth-pdb --sequence "LKELEKELEKELEKELEKELEKEL" \
--conformation alpha \
--minimize \
--gen-nef --noe-cutoff 6.0 \
--gen-relax --field 800 \
--visualize
```
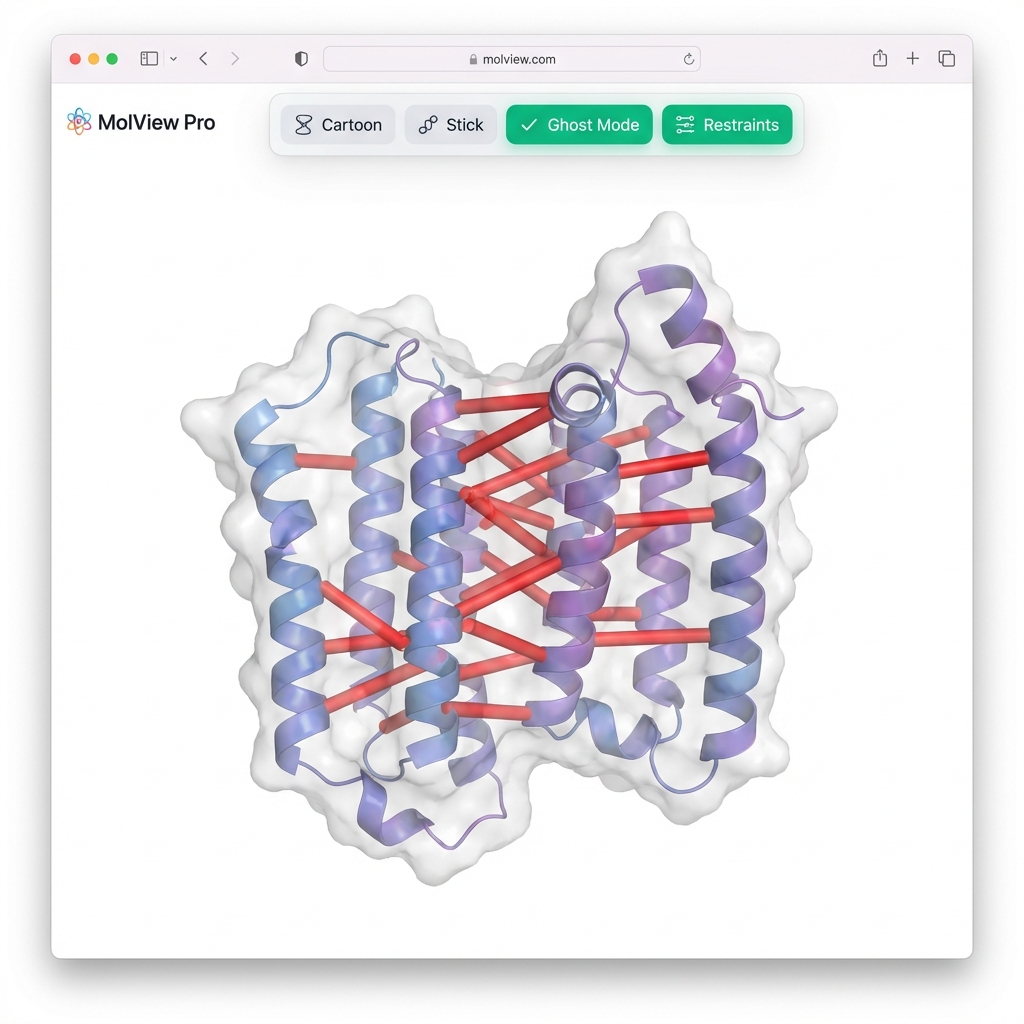
> **👀 Viewer Tip**: Since you used `--gen-nef`, the **synthetic NOE restraints** will automatically appear as **red cylinders** connecting the protons. Use the **"🔴 Restraints"** button in the viewer to toggle them on/off!
#### 🌿 Amphipathic Helix Visualization
A classic biophysical motif where one face of the helix is hydrophobic (L, V, I) and the other is hydrophilic (K, E, R).
```bash
# Generate and Minimize
synth-pdb --sequence "LKWLKRLLKWLKRLLKWLKRL" --conformation alpha --minimize --visualize
```
*In the viewer*: Switch to **"Sphere"** style and **"Element"** color. You will see the "greasy" hydrophobic patch (Carbon-rich) clearly separated from the charged residues (Nitrogen/Oxygen-rich). This "hydrophobic moment" drives membrane binding!
> **🎓 Academic Note - "Amphipathic"**:
> From Greek *amphi* (both) and *pathos* (feeling). An amphipathic helix has a "split personality":
> * **Hydrophobic Face** (L, V, I, F): Hates water. Buries itself inside the protein core or membrane.
> * **Hydrophilic Face** (K, R, E, D): Loves water. Faces the solvent to keep the protein soluble.
> This duality is the fundamental force driving protein folding! 🧬🌗
## Validation & Refinement
### Validation Checks
When `--validate` is enabled, the tool checks for:
1. **Bond Lengths**: Compares N-CA, CA-C, C-N, C-O distances against standard values (±0.05 Å tolerance)
2. **Bond Angles**: Validates N-CA-C, CA-C-N, CA-C-O angles (±5° tolerance)
3. **Ramachandran Angles**: Checks phi/psi dihedral angles against MolProbity-defined polygonal regions
- **Categories**: General, Glycine, Proline, Pre-Proline
- **Levels**: Distinguishes between Favored, Allowed, and Outlier status
4. **Steric Clashes**: Detects atoms that are too close
- Minimum distance rule: ≥2.0 Å between any atoms
- van der Waals overlap: atoms closer than sum of vdW radii
5. **Peptide Plane Planarity**: Checks omega (ω) dihedral angles
- Trans: ~180° (±30° tolerance)
- Cis: ~0° (±30° tolerance)
6. **Sequence Improbabilities**: Flags unusual sequence patterns
- Charge clusters (4+ consecutive charged residues)
- Long hydrophobic stretches (8+ residues)
- Odd cysteine counts (unpaired cysteines)
- Poly-proline or poly-glycine runs
7. **Chirality**: Validates L-amino acid stereochemistry
- Checks improper dihedral angle N-CA-C-CB
- L-amino acids should have proper chirality (improper dihedral ±60° to ±120°)
- Glycine is automatically exempt (no CB atom)
- Detects incorrect stereochemistry (D-amino acids)
### Refinement Strategy
The `--refine-clashes` option uses an iterative approach:
1. Identifies clashing atom pairs
2. Slightly adjusts positions to increase separation
3. Re-validates structure
4. Stops when no improvement or max iterations reached
> **Note**: Refinement focuses on steric clashes and may introduce other violations. Use in combination with `--best-of-N` for better results.
## Output PDB Format
### Structure Representation
- **Full Atomic Model**: All backbone atoms (N, CA, C, O) + side-chain heavy atoms + hydrogens
- **Geometry**: Linear alpha-helix conformation along the X-axis
- **Chain ID**: Always 'A'
- **Residue Numbering**: Sequential from 1
- **Terminal Modifications**: N-terminal and C-terminal hydrogens/oxygens included
### Atomic Records & B-Factors
Each atom line follows the standard PDB format. The **B-factor** (Temperature Factor) is stored in **columns 61-66**.
```text
ATOM 1 N ALA A 1 -2.193 1.858 1.271 0.85 56.71 N
ATOM 5 CB ALA A 1 0.241 1.845 1.013 0.85 86.14 C
^^^^ ^^^^^
Occpy B-Fact
```
* **Occupancy (0.85)**: Reflects the Order Parameter ($S^2$) if calculated, or default.
* **B-Factor (56.71 vs 86.14)**: Reflects atomic mobility. Note how the side-chain atom (CB) has a higher B-factor than the backbone (N), indicating greater flexibility.
### Header Information
Generated PDB files include standard header records:
```
HEADER PEPTIDE
TITLE GENERATED LINEAR PEPTIDE OF LENGTH
REMARK 1 This PDB file was generated by the CLI 'synth-pdb' tool.
REMARK 2 It represents a simplified model of a linear peptide chain.
REMARK 2 Coordinates are idealized and do not reflect real-world physics.
REMARK 3 GENERATION PARAMETERS:
REMARK 3 Command: synth-pdb --length 10 --validate ...
```
The **REMARK 3** records store the exact command-line arguments used for **reproducibility**.
### Validation Reports
When `--validate` is used, violations are reported:
```
WARNING --- PDB Validation Report for /path/to/file.pdb ---
WARNING Final PDB has 5 violations.
WARNING Bond length violation: N-1-A to CA-1-A. Distance: 1.52Å, Expected: 1.46ű0.05Å
WARNING Steric clash (min distance): Atoms CA-3-A and CB-3-A are too close (1.85Å)...
```
## Scientific Context
### Intended Use Cases
✅ **Appropriate Uses:**
- Testing PDB parsers and file I/O
- Benchmarking structure validation tools
- Educational demonstrations of protein structure concepts
- Generating test datasets for bioinformatics pipelines
- Placeholder structures for software development
❌ **Inappropriate Uses:**
- Homology modeling templates
- Drug docking studies
- Experimental predictions
- Publication-quality structures
Real protein structures require sophisticated methods like:
- Molecular dynamics with force fields (AMBER, CHARMM)
- Quantum mechanics calculations (DFT)
- Energy minimization and conformational search
- Crystallographic or NMR experimental data
## Limitations
### Structural Limitations
1. **Topology**:
- Primarily generates **linear** variations or simple **disulfide-bonded** loops.
- Does not perform *de novo* folding (prediction of tertiary structure from sequence).
- Multi-chain complexes are currently limited to simple docking preparations.
2. **Geometry**:
- **Default Mode**: Uses idealized internal coordinates (perfect bond lengths/angles).
- **Physically Realistic Mode** (`--minimize`): Resolves this by relaxing the structure with OpenMM, but is computationally more expensive.
3. **Rotamer Library**:
- **Backbone-Dependent**: Fully implemented for **All 20 Amino Acids**.
- **Mechanism**: Checks local secondary structure (Alpha/Beta) to select rotamers that avoid backbone clashes.
- **Rare Rotamers**: Very rare side-chain conformations (<1% probability) may be undersampled.
4. **Environmental Effects**:
- **Solvent**: Uses Implicit Solvent (OBC2) to model water screening, but lacks explicit water molecules.
- **Membranes**: No lipid bilayer simulation for transmembrane proteins.
### Validation Limitations
- **Ramachandran Regions**: Uses simplified **rectangular boundaries** for valid phi/psi regions. While faster, this is less rigorous than the contoured probability density functions used by MolProbity.
- **Electrostatics**: Basic clash detection does not account for long-range electrostatic repulsion/attraction (though `--minimize` does).
- **Protonation**: Simple pH-based titration (His/Asp/Glu) without full pKa calculation.
### Terminology: Decoys vs NMR Ensembles
There is an important distinction between the "Decoys" generated by this tool and a traditional "NMR Ensemble":
* **NMR Ensemble**: A set of structures (usually 20) that *all satisfy* experimental restraints (NOEs) and have converged to the same fold. They represent the **precision** of the structure determination.
* **Decoys (Conformational Ensemble)**: A set of independent structures generated to sample the conformational space. They often have high RMSD (diversity) and represent the **search space**.
`synth-pdb --mode decoys` generates the latter: independent snapshots. To create a pseudo-NMR ensemble, use `--rmsd-max 2.0` to filter for similar structures.
### Performance Considerations
- `--guarantee-valid` may **never converge** for long sequences (>50 residues)
- Combinatorial explosion of possible violations
- Consider using `--best-of-N` instead
- `--refine-clashes` is **iterative and may be slow** for large structures
- Each iteration requires full re-validation
- Validation runtime scales with sequence length (O(N²) for steric clashes)
## Development
### Running Tests
```bash
# All tests
pytest -v
# With coverage
pytest --cov=synth_pdb --cov-report=term-missing
# Specific test file
pytest tests/test_generator.py -v
```
**Test Coverage**: 93% overall
- 1369 tests covering generation, validation, CLI and edge cases
### Project Structure
```
synth-pdb/
├── synth_pdb/
│ ├── __init__.py
│ ├── main.py # CLI entry point
│ ├── generator.py # PDB structure generation (NeRF, rotamers, PTMs, D-AAs)
│ ├── validator.py # Validation checks & get_quality_report()
│ ├── physics.py # OpenMM energy minimization, MD, simulate_trajectory()
│ ├── data.py # Constants, rotamer library, Ramachandran polygons
│ ├── nmr.py # RPF scores, NOE compatibility shims (delegates to synth-nmr)
│ ├── rdc.py # Residual Dipolar Coupling (Saupe-matrix formalism)
│ ├── msa.py # MCMC Potts-model MSA co-evolution generator
│ ├── plm.py # ESM-2 protein language model embeddings
│ ├── orientogram.py # 6D rotation-invariant inter-residue orientation
│ ├── batch_generator.py # Vectorized BatchedGenerator for AI training
│ ├── decoys.py # Hard-decoy generation (threading, drift, shuffle)
│ ├── dataset.py # Bulk dataset generation (NPZ / PDB format)
│ ├── chemical_shifts.py # SPARTA-lite + ring-current shift prediction
│ ├── biophysics.py # Biophysical utility functions
│ ├── viewer.py # 3Dmol.js browser-based visualizer
│ ├── geometry/ # Geometry subpackage (v1.27+)
│ │ ├── superposition.py # Kabsch algorithm, apply_transformation, find_medoid
│ │ ├── rmsd.py # RMSD, pairwise RMSD, symmetry-aware variants
│ │ ├── dihedral.py # Dihedral angle calculations
│ │ ├── nerf.py # NeRF backbone construction kernels
│ │ ├── sidechain.py # Side-chain geometry helpers
│ │ └── vectorized.py # NumPy-vectorized / Numba-JIT geometry kernels
│ ├── ensemble/ # NMR ensemble analysis subpackage (v1.34.1+)
│ │ ├── daop.py # DAOPCalculator (Hyberts 1992 dihedral order parameters)
│ │ └── statistics.py # EnsembleStatistics, QualityAssessment dataclasses
│ └── quality/ # Structure quality scoring (v1.18+)
│ ├── gnn/ # Graph Neural Network quality scorer
│ ├── classifier.py # Random Forest / GNN quality filter interface
│ └── features.py # Feature extraction for quality models
├── tests/
│ ├── test_generator.py
│ ├── test_validator.py
│ ├── test_scientific_validation.py
│ ├── test_coupling.py
│ ├── unit/ # Unit tests for geometry, ensemble, quality modules
│ └── ... (many more)
├── examples/
│ ├── interactive_tutorials/
│ ├── ml_integration/
│ └── ml_loading/ # JAX / PyTorch / MLX zero-copy handover
├── docs/
├── incubator/
├── pyproject.toml
└── README.md
```
## 📚 Biophysical References & Further Reading
For students and researchers interested in the physics behind the code, here are key seminal papers:
* **Cis-Proline (~5% Frequency):**
* MacArthur, M. W., & Thornton, J. M. (1991). Influence of proline residues on protein conformation. *J Mol Biol*, 218(2), 397-412.
* Weiss, M. S., et al. (1998). Cis-proline. *Acta Cryst D*, 54, 323-329.
* **Macrocyclization & Cyclic Peptides:**
* Horton, D. A., et al. (2003). The combinatorial synthesis of bicyclic peptides. *Chem. Rev.*, 103(3), 893-930. (Seminal review on macrocycles).
* Craik, D. J., et al. (2013). The future of peptide-based drugs. *Chem. Biol. Drug Des.*, 81(1), 136-147.
* **NMR Structure Validation & Chirality:**
* Montelione, G. T., et al. (2013). Recommendations of the wwPDB NMR Validation Task Force. *Structure*, 21(9), 1563-1570. (Defines standards for geometric validation).
* Huang, Y. J., Powers, R., & Montelione, G. T. (2005). "Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics." *Journal of the American Chemical Society*, 127(6), 1665-1674.
* Raman, S., et al. (2010). "NMR Structure Determination for Larger Proteins Using Backbone-Only Data." *Science*, 327(5968), 1014-1018. (Using chemical shifts and RDCs for structure prediction).
* Bhattacharya, A., & Montelione, G. T. (2011). PDBStat: a server for validation of protein NMR structures.
* **Nuclear Overhauser Effect (NOE) & $r^{-6}$:**
* Wüthrich, K. (1986). *NMR of Proteins and Nucleic Acids*. Wiley-Interscience. (The definitive text).
* Wüthrich, K. (2003). Nobel Lecture: NMR Studies of Protein Structure and Dynamics.
* **Chemical Shift Prediction (SPARTA) & Referencing (DSS):**
* Shen, Y., & Bax, A. (2010). SPARTA+: a modest improvement in empirical NMR chemical shift prediction... *J Biomol NMR*, 48, 13-22.
* Markley, J. L., et al. (1998). Recommendations for the presentation of NMR structures... (IUPAC). *Pure Appl Chem*, 70(1), 117-142. (Defined DSS as the standard).
* **Internal Dynamics & Model-Free Formalism:**
* Lipari, G., & Szabo, A. (1982). Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. *J. Am. Chem. Soc.*, 104(17), 4546–4559. (The foundational theory).
* Kay, L. E., Torchia, D. A., & Bax, A. (1989). Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy... *Biochemistry*, 28(23), 8972-8979. (The seminal application to proteins).
## References & Bibliography
### Structure Generation & Rotamers
1. **Dunbrack, R. L., & Cohen, F. E. (1997).** Bayesian statistical analysis of protein side-chain rotamer preferences. *Protein Science, 6*(8), 1661–1681.
- Used for: Rotamer libraries and side-chain probability distributions.
2. **Parsons, J., et al. (2005).** Practical conversion from torsion space to Cartesian space for in silico protein synthesis. *Journal of Computational Chemistry, 26*(10), 1063–1068.
- Used for: The NeRF (Natural Extension Reference Frame) algorithm for backbone construction.
3. **MacArthur, M. W., & Thornton, J. M. (1991).** Influence of proline residues on protein conformation. *Journal of Molecular Biology*, 218(2), 397-412.
- Used for: Cis-Proline isomerization statistics (~5% cis frequency).
4. **Homeyer, N., et al. (2006).** AMBER force-field parameters for phosphorylated amino acids... *Journal of Molecular Modeling*, 12(3), 281-289.
- Used for: PTM physics parameters (SEP, TPO, PTR) in OpenMM.
5. **Smith, D. M. (2001).** Protein Composition and Structure. *Encyclopedia of Life Sciences*.
- Used for: Biological amino acid frequency data.
### NMR Dynamics & Relaxation
6. **Lipari, G., & Szabo, A. (1982).** Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. *Journal of the American Chemical Society, 104*(17), 4546–4559.
- Used for: Calculating $S^2$ order parameters and relaxation rates ($R_1, R_2, NOE$).
7. **Wishart, D. S., et al. (1995).** 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. *Journal of Biomolecular NMR, 6, 135–140.*
- Used for: Random coil chemical shift baselines.
8. **Cavanagh, J., et al. (2007).** *Protein NMR Spectroscopy: Principles and Practice*. Academic Press.
- Used for: General NMR theory and relaxation equations.
### Validation
7. **Williams, C. J., et al. (2018).** MolProbity: More and better reference data for improved all-atom structure validation. *Protein Science, 27*(1), 293–315.
- Used for: Ramachandran polygon definitions and validation criteria.
8. **Lovell, S. C., et al. (2003).** Structure validation by Calpha geometry: phi,psi and Cbeta deviation. *Proteins: Structure, Function and Bioinformatics, 50*(3), 437–450.
- Used for: Early reference for Ramachandran validation concepts.
## Glossary of Scientific Terms & Acronyms
This section provides definitions and seminal references for the biophysical and computational terms used throughout `synth-pdb`. Entries are sorted alphabetically.
| Term | Definition | Reference |
| :--- | :--- | :--- |
| **AMBER** | **Assisted Model Building with Energy Refinement**. A widely-used suite of molecular simulation programs and force fields for biomolecules. | Case, D. A., et al. (2005). *J. Comput. Chem.* |
| **B-factor** | **Temperature Factor** (8π²⟨u²⟩). Measures atomic displacement due to thermal motion and static disorder. Higher values indicate greater flexibility; lower values indicate rigidity. | — |
| **Backbone-Dependent Rotamer** | A side-chain conformation probability that depends on the local backbone angles (φ, ψ). Used to select realistic side-chain orientations based on secondary structure context. | Dunbrack & Cohen (1997). *Protein Science.* |
| **CASP** | **Critical Assessment of Structure Prediction**. A community-wide experiment held every two years to establish the state-of-the-art in protein structure modeling. | Kryshtafovych, A., et al. (2021). *Proteins.* |
| **Chi Angles (χ)** | Dihedral angles describing side-chain conformation about successive bonds from Cα outward (χ₁, χ₂, …). Discrete preferred values define rotamers. | — |
| **CSI** | **Chemical Shift Index**. A standard method used to deduce protein secondary structure (alpha helix vs. beta sheet) from detected NMR chemical shift deviations. | Wishart, D. S., et al. (1992). *Biochemistry.* |
| **Macrocycle** | A cyclic macromolecule or macromolecular network, such as a cyclic peptide or a crown ether. In therapeutic chemistry, macrocyclization improves metabolic stability and binding affinity. | IUPAC Gold Book. |
| **MolProbity** | A structure validation web service and scoring function providing the gold standard for Ramachandran and rotamer analysis. | Chen, V. B., et al. (2010). *Acta Cryst. D.* |
| **NEF** | **NMR Exchange Format**. A unified, open standard for the exchange of NMR restraint data among various software packages. | Gutmanas, A., et al. (2015). *Nat. Struct. Mol. Biol.* |
| **NeRF** | **Natural Extension Reference Frame**. An algorithm for rapidly constructing 3D Cartesian coordinates from internal coordinates (bond lengths, angles, and dihedrals). | Parsons, J., et al. (2005). *J. Comput. Chem.* |
| **NOE** | **Nuclear Overhauser Effect**. A phenomenon where magnetization is transferred between spins through space, allowing measurement of inter-atomic distances (r⁻⁶ dependency). | Wüthrich, K. (1986). *NMR of Proteins and Nucleic Acids.* |
| **OBC2** | **Onufriev-Bashford-Case model 2**. A computationally efficient implicit solvent model (Generalized Born) used to simulate the screening effect of water on charged groups. | Onufriev, A., et al. (2004). *Proteins.* |
| **PDB** | **Protein Data Bank**. The global repository for 3D structural data of proteins, nucleic acids, and complex assemblies. | Berman, H. M., et al. (2000). *Nucleic Acids Res.* |
| **Phi/Psi (φ, ψ)** | Backbone dihedral angles. φ is defined by C(i−1)−N−Cα−C; ψ is defined by N−Cα−C−N(i+1). Together they determine backbone geometry and are plotted on the Ramachandran plot. | — |
| **Pre-Proline** | The residue immediately preceding a Proline. It has restricted conformational freedom due to steric clash with the Proline ring, and uses a distinct Ramachandran distribution. | — |
| **Ramachandran Plot** | A 2D plot of φ vs ψ angles showing energetically allowed and disallowed backbone conformations for amino acids. The basis for structural validation. | Ramachandran et al. (1963). *J. Mol. Biol.* |
| **Rotamer** | Short for "Rotational Isomer". Preferred, low-energy side-chain conformations defined by discrete χ-angle clusters. | Dunbrack, R. L. (2002). *Curr. Opin. Struct. Biol.* |
| **S²** | **Model-Free Order Parameter** (Lipari-Szabo). A value between 0 (random/flexible) and 1 (perfectly rigid) describing the degree of spatial restriction of local backbone motion on ps–ns timescales. | Lipari, G., & Szabo, A. (1982). *J. Am. Chem. Soc.* |
| **SASA** | **Solvent Accessible Surface Area**. The surface area of a biomolecule accessible to a solvent probe (typically a 1.4 Å water molecule). Low SASA indicates a buried residue; high SASA indicates solvent exposure. | Shrake & Rupley (1973). *J. Mol. Biol.* |
| **BMRB** | **BioMagResBank**. The international repository for NMR spectroscopic data derived from biological molecules, including chemical shift assignments, restraint files, and relaxation data. | Ulrich, E. L., et al. (2008). *Nucleic Acids Res.* |
| **DAOP** | **Dihedral Angle Order Parameter**. A circular statistics metric (range 0–1) quantifying the consistency of backbone dihedral angles (φ, ψ) across an NMR ensemble. Well-defined residues satisfy S(φ)+S(ψ) ≥ 1.8 (PDBStat convention). Available via `synth_pdb.ensemble.daop`. | Hyberts, S. G., et al. (1992). *Protein Science* 1:736. |
| **DCA** | **Direct Coupling Analysis**. A statistical inference method that identifies evolutionarily co-varying residue pairs in a multiple sequence alignment to predict spatial contacts and generate AlphaFold-ready MSA inputs. | Morcos, F., et al. (2011). *PNAS* 108:E1293. |
| **Engh & Huber** | The landmark (1991) set of ideal bond lengths and bond angles for the 20 standard amino acids, derived from small-molecule crystallography. `PDBValidator` uses these as Z-score reference distributions (v1.29+). | Engh, R. A., & Huber, R. (1991). *Acta Cryst. A* 47:392. |
| **ESM-2 / PLM** | **Evolutionary Scale Modeling 2 / Protein Language Model**. A large transformer trained on millions of protein sequences that produces per-residue embeddings for zero-shot quality scoring. Available via `synth_pdb.quality.plm`; install with `pip install synth-pdb[plm]`. | Lin, Z., et al. (2023). *Science* 379:1123. |
| **GNN** | **Graph Neural Network**. A deep learning model operating on graph-structured data. In `synth_pdb.quality.gnn`, residues are nodes and spatial/sequence contacts are edges, enabling structure quality assessment. Install with `pip install synth-pdb[gnn]`. | Kipf, T. N., & Welling, M. (2017). *ICLR.* |
| **IDR / IDP** | **Intrinsically Disordered Region / Protein**. A protein region that lacks a stable 3D fold under physiological conditions. Characterised by high RMSF, low S², and low AlphaFold pLDDT. Validated against PRE NMR data in `idp_ensemble_validation.ipynb`. | Dyson, H. J., & Wright, P. E. (2005). *Nat. Rev. Mol. Cell Biol.* |
| **Kauzmann (Hydrophobic Effect)** | The thermodynamic driving force for hydrophobic residues to bury in a protein's core, arising from the entropic cost of ordering water around non-polar groups. Cited in SASA burial validation (v1.29). | Kauzmann, W. (1959). *Adv. Protein Chem.* 14:1. |
| **Magic Step** | A coupled MCMC mutation proposal in the MSA Potts-Model sampler where two spatially contacting residues are mutated simultaneously, preserving co-evolutionary constraints (20% proposal rate, v1.26+). | — |
| **MCMC / Metropolis-Hastings** | **Markov Chain Monte Carlo**. A class of algorithms for sampling from probability distributions. Used in `synth_pdb.msa` to simulate protein sequence evolution on the Potts Model energy landscape. | Metropolis, N., et al. (1953). *J. Chem. Phys.* 21:1087. |
| **Orientogram** | A 6D rotation-invariant representation of inter-residue orientations in a protein structure, used as a structural fingerprint and neural network input feature. See `synth_pdb.orientogram`. | — |
| **pLDDT** | **Predicted Local Distance Difference Test**. AlphaFold2's per-residue confidence score (0–100). Low pLDDT (< 50) accurately signals intrinsically disordered regions — not prediction failure. Correlates inversely with NMR S² and MD RMSF. | Jumper, J., et al. (2021). *Nature* 596:583. |
| **Potts Model** | A statistical physics model of interacting spins on a lattice, applied in `synth_pdb.msa` to protein sequences: each position is a spin (amino acid) and J_ij couplings encode co-evolutionary interactions between residue pairs. | Weigt, M., et al. (2009). *PNAS* 106:67. |
| **PPII** | **Polyproline II Helix**. A left-handed helical conformation (φ ≈ −75°, ψ ≈ +145°) common in collagen and proline-rich sequences. Specifiable via `--conformation ppii`. | — |
| **PRE** | **Paramagnetic Relaxation Enhancement**. An NMR phenomenon where a paramagnetic spin label broadens nearby nuclear resonances proportional to r⁻⁶. Used to validate IDP conformational ensembles. | Clore, G. M., & Iwahara, J. (2009). *Chem. Rev.* 109:4108. |
| **Q-factor** | A dimensionless goodness-of-fit metric for Residual Dipolar Couplings: Q = RMSD(D_calc − D_obs) / RMSD(D_obs). Lower is better; high-quality structures typically achieve Q < 0.20. | Cornilescu, G., et al. (1998). *J. Biomol. NMR* 12:373. |
| **RDC** | **Residual Dipolar Coupling**. An NMR observable arising when a molecule is partially aligned in an anisotropic medium. Encodes long-range bond-vector orientation information relative to the molecular alignment frame. Computed by `synth_pdb.rdc`. | Tjandra, N., & Bax, A. (1997). *Science* 278:1111. |
| **RMSF** | **Root Mean Square Fluctuation**. The standard deviation of each residue's position over time in an MD trajectory (after Kabsch rigid-body alignment). High RMSF = flexibility; Low RMSF = rigidity. Inversely related to S² and pLDDT. | — |
| **Saupe Matrix / Alignment Tensor** | The 3×3 traceless symmetric tensor describing the degree and orientation of molecular alignment in an anisotropic medium. Parameterised by axial component `Da` and rhombicity `R` for RDC calculations. | Saupe, A. (1968). *Angew. Chem.* 7:97. |
| **Top2018** | A high-resolution Ramachandran reference dataset derived from ~15,000 protein chains (resolution < 1.5 Å), superseding Top8000. Adopted in `PDBValidator` from v1.29 for more accurate φ/ψ boundary validation. | — |
| **Top8000** | A high-quality curated dataset of ~8000 protein chains (resolution < 2.0 Å, low sequence homology) used to derive accurate Ramachandran contours and rotamer libraries. | Lovell, S. C., et al. (2003). *Proteins.* |
## License
This project is provided as-is for educational and testing purposes.
---
## Citation
If you use this software in your research, please cite:
```bibtex
@software{synth_pdb,
author = {Elkins, George},
title = {synth-pdb: Realistic Protein Structure Generator},
year = {2026},
url = {https://github.com/elkins/synth-pdb}
}
```
## 🛠️ Software & Libraries
This project relies on the following open-source scientific software:
- **[OpenMM](https://openmm.org/)**: High-performance molecular dynamics toolkit used for physics-based energy minimization (Implicit Solvent/OBC2).
- **[Biotite](https://www.biotite-python.org/)**: Comprehensive library for structural biology involved in PDB IO, atom manipulation, and geometric analysis.
- **[3Dmol.js](https://3dmol.csb.pitt.edu/)**: JavaScript library for molecular visualization used in the `--visualize` browser-based viewer.
- **[NumPy](https://numpy.org/)**: Fundamental package for scientific computing and matrix operations.
### Tools with NEF Support
These external tools can import the data generated by `synth-pdb`:
- **[CCPNMR Analysis](https://ccpn.ac.uk/)**: Premier software for NMR data analysis, assignment, and structure calculation (Native NEF support).
- **[CYANA](http://www.cyana.org/)**: Automated NMR structure calculation.
- **[XPLOR-NIH](https://nmr.cit.nih.gov/xplor-nih/)**: Biomolecular structure determination.
## 📚 References & Scientific Publications
### Key Publications in NMR Structure Validation
1. **Protein Structure Validation Suite (PSVS)**
* Bhattacharya, A., Tejero, R., & Montelione, G. T. (2007). "Evaluating protein structures determined by structural genomics consortia." *Proteins: Structure, Function, and Bioinformatics*, 66(4), 778-795.
* [Link to Publisher](https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.21165)
2. **RPF Scores (Recall, Precision, F-measure)**
* Huang, Y. J., Powers, R., & Montelione, G. T. (2005). "Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics." *Journal of the American Chemical Society*, 127(6), 1665-1674.
* [Link to Publisher](https://pubs.acs.org/doi/10.1021/ja0471963)
3. **DP Score (Discriminant Power)**
* Huang, Y. J., Tejero, R., Powers, R., & Montelione, G. T. (2006). "A topology-constrained distance network algorithm for protein structure determination from NOESY data." *Proteins: Structure, Function, and Bioinformatics*, 62(3), 587-603.
* [Link to Publisher](https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.20784)
### Data Standards
- **NMR Exchange Format (NEF)**
* Gutmanas, A., et al. (2015). "NMR Exchange Format: a unified and open standard for representation of NMR restraint data." *Nature Structural & Molecular Biology*, 22, 433–434.
* [Link to Publisher](https://www.nature.com/articles/nsmb.3041)
* **Extension Proposal:** "Proposal For Incorporating NMR Relaxation Data In NEF" (GitHub PDF)
* [Link to Proposal](https://github.com/NMRExchangeFormat/NEF/blob/master/specification/Proposal