https://github.com/fgvieira/ngsld
Calculation of pairwise Linkage Disequilibrium (LD) under a probabilistic framework
https://github.com/fgvieira/ngsld
genotype-likelihoods ld ngs population-genomics
Last synced: 11 months ago
JSON representation
Calculation of pairwise Linkage Disequilibrium (LD) under a probabilistic framework
- Host: GitHub
- URL: https://github.com/fgvieira/ngsld
- Owner: fgvieira
- License: gpl-2.0
- Created: 2015-10-31T15:43:49.000Z (over 10 years ago)
- Default Branch: master
- Last Pushed: 2023-11-13T15:24:14.000Z (over 2 years ago)
- Last Synced: 2023-11-13T16:34:13.937Z (over 2 years ago)
- Topics: genotype-likelihoods, ld, ngs, population-genomics
- Language: C++
- Homepage:
- Size: 301 KB
- Stars: 33
- Watchers: 3
- Forks: 7
- Open Issues: 10
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
README
# ngsLD
`ngsLD` is a program to estimate pairwise linkage disequilibrium (LD) taking the uncertainty of genotype's assignation into account. It does so by avoiding genotype calling and using genotype likelihoods or posterior probabilities.
### Citation
`ngsLD` has been published in [Bioinformatics](https://doi.org/10.1093/bioinformatics/btz200), so please cite it if you use it in your work:
Fox EA, Wright AE, Fumagalli M, and Vieira FG
ngsLD: evaluating linkage disequilibrium using genotype likelihoods
Bioinformatics (2019) 35(19):3855 - 3856
### Installation
`ngsLD` can be easily installed but has some external dependencies:
* Mandatory:
* `gcc`: >= 4.9.2 tested on Debian 7.8 (wheezy)
* `zlib`: v1.2.7 tested on Debian 7.8 (wheezy)
* `gsl` : v1.15 tested on Debian 7.8 (wheezy)
* Optional (only needed for testing or auxilliary scripts):
* `md5sum`
* `Perl` packages: `Getopt::Long`, `Graph::Easy`, `Math::BigFloat`, and `IO::Zlib`
* `R` packages: `optparse`, `tools`, `ggplot2`, `reshape2`, `plyr`, `gtools`, and `LDheatmap`
* `python3` packages: `graph-tool`, and `pandas`
To install the entire package just download the source code:
% git clone https://github.com/fgvieira/ngsLD.git
and run:
% cd ngsLD
% make
To run the tests (only if installed through [ngsTools](https://github.com/mfumagalli/ngsTools)):
% make test
Executables are built into the main directory. If you wish to clean all binaries and intermediate files:
% make clean
### Usage
% ./ngsLD [options] --geno glf/in/file --n_ind INT --n_sites INT --out output/file
#### Parameters
* `--geno FILE`: input file with genotypes, genotype likelihoods or genotype posterior probabilities.
* `--probs`: is the input genotype probabilities (likelihoods or posteriors)?
* `--log_scale`: is the input in log-scale?
* `--n_ind INT`: sample size (number of individuals).
* `--n_sites INT`: total number of sites.
* `--pos(H)` FILE: input file with site coordinates (one per line), where the 1st column stands for the chromosome/contig and the 2nd for the position (bp); remaining columns will be ignored but included in output; `--posH` assumes there is a header.
* `--max_kb_dist DOUBLE`: maximum distance between SNPs (in Kb) to calculate LD. Set to `0`(zero) to disable filter. [100]
* `--max_snp_dist INT`: maximum distance between SNPs (in number of SNPs) to calculate LD. Set to `0` (zero) to disable filter. [0]
* `--min_maf DOUBLE`: minimum SNP minor allele frequency. [0]
* `--ignore_miss_data`: ignore missing genotype data from analyses (e.g. MAF and haplotype frequency estimation).
* `--call_geno`: call genotypes before running analyses.
* `--N_thresh DOUBLE`: minimum threshold to consider position; if highest GL is lower, set as missing data (assumes -call_geno).
* `--call_thresh DOUBLE`: minimum threshold to call genotype; if highest GL is lower, left as is (assumes -call_geno).
* `--rnd_sample DOUBLE`: proportion of comparisons to randomly sample. [1]
* `--seed INT`: random number generator seed for random sampling (--rnd_sample).
* `--extend_out`: print extended output (see below).
* `--out FILE`: output file name. [stdout]
* `--n_threads INT`: number of threads to use. [1]
* `--verbose INT`: selects verbosity level. [1]
### Input data
As input, `ngsLD` accepts both genotypes, genotype likelihoods (GP) or genotype posterior probabilities (GP). Genotypes must be input as gziped TSV with one row per site and one column per individual ) and genotypes coded as [-1, 0, 1, 2].
As for GL and GP, `ngsLD` accepts both gzipd TSV and binary formats, but with 3 columns per individual ) and, in the case of binary, the GL/GP coded as doubles.
It is advisable that SNPs be called first, since monomorphic sites are not informative and it will greatly speed up computation. If not, these comparisons will show up as `nan` or `inf` in the output.
### Output
`ngsLD` outputs a TSV file with LD results for all pairs of sites for which LD was calculated, where the first two columns are positions of the SNPs, the third column is the distance (in bp) between the SNPs, and the following 4 columns are the various measures of LD calculated (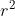 from pearson correlation between expected genotypes, 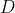 from EM algorithm, 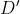 from EM algorithm, and 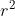 from EM algorithm). If the option `--extend_out` is used, then an extra 8 columns are printed with number of samples, minor allele frequency (MAF) of both loci, haplotype frequencies for all four haplotypes, and a chi2 (1 d.f.) for the strength of association ([Collins et al., 1999](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC24792/)).
If both `--max_kb_dist` and `--max_snp_dist` are set to `0`, `ngsLD` will output all comparisons, even between different chromosomes/scaffolds/contigs.
### Possible analyses
##### LD pruning
For some analyses, linked sites are typically pruned since their presence can bias results. You can use [prune_graph](https://github.com/fgvieira/prune_graph) to prune your dataset and get a list of unlinked sites. Alternatively, you can also use the auxiliary scripts `scripts/prune_graph.pl` or `scripts/prune_ngsLD.py`, but they are much slower (specially the perl script) and no longer supported.
```
prune_graph --header --in testLD_2.ld --weight-field "r2" --weight-filter "dist <= 50000 && r2 >= 0.5" --out testLD_unlinked.pos
```
For more advanced options, please check help (`prune_graph --help`).
NOTE: as of version `1.2.1` the MAF filtering has been disabled in `ngsLD`, due to potential misleading prunning results. Briefly, filtered sites due to low MAF are skipped by `ngsLD` and not included in the output file. As such, when pruning, if the output list of unlinked sites is used to extract independent sites from a dataset, all low MAF sites will also be excluded (since they were not in the `ngsLD` output). If you want to include them, either perform the MAF filtering when prunning (you need `ngsLD` extended output) or use the list of linked sites and exclude them from your dataset.
#### LD decay
If you are interested on the rate of LD decay, you can fit a distribution to your data using the script `scripts/fit_LDdecay.R` to fit LD decay models for 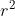 ([Hill and Weir, 1988](https://www.ncbi.nlm.nih.gov/pubmed/3376052) and [Remington et al., 2001](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC58755/)) and 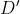 ([Abecassis et al., 2001](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1234912/)) over physical (or genetic) distance.
There are two models implemented for 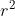 and one for 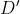 decay. Briefly, the first is derived by adjusting the expected 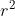 under a drift-recombination equilibrium for finite samples sizes and low level of mutation ([Hill and Weir, 1988](https://www.ncbi.nlm.nih.gov/pubmed/3376052)) with a single parameter (rate of decay):
(11+C)}\right]\cdot\left[1+\frac{(3+C)(12+12C+C^2)}{n(2+C)(11+C)}\right])
The second formulation is an extension of the [Sved, 1971](https://www.ncbi.nlm.nih.gov/pubmed/5170716) model (expected 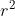 under drift-recombination equilibrium) with three parameters (rate of decay, maximum observed 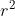 and minimum observed 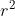) to account for the range of observed 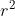 values:

where the rate of decay .
For 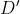, we fit the expectation derived by [Abecassis et al., 2001](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1234912/), assuming a recombination rate of 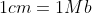 and fixing , resulting also on a three parameter model (rate of decay, maximum 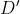 and minimum 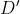):
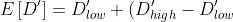\cdot&space;D'_0&space;\cdot&space;(1-\theta)^t)
For more information, please refer to the online published supplementary data.
% Rscript --vanilla --slave scripts/fit_LDdecay.R --ld_files ld_files.list --out plot.pdf
* `--ld_files FILE`: list of LD files to fit and plot (if ommited, can be read from STDIN)
* `--out`: Name of output plot
* `--n_ind`: Only relevant when fitting 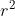 decay to specify the first model
For more advanced options, please check script help (`Rscript --vanilla --slave scripts/fit_LDdecay.R --help`). The shape of the decay curve can give valuable insight into the sample's biology (e.g.):
* `Intersect` (high): small Ne (or bottleneck), natural selection (local LD)*, non-random mating (or inbreeding), recent admixture
* `Decay Rate` (high): large Ne (or population expansion)*, high mutation rate*, high recombination rate*, random mating (or no inbreeding)*, not recently admixture
* `Asymptote` (high): population structure*, small sample size*
NOTE: Please keep in mind that these are just general trends, and that it all depends on the biology/genetics of the sample. Since LD is influenced by many factors, it is usually less straightforward to derive exact predictions from it.
##### LD blocks
To plot LD blocks, we also provide a small script as an example for how it can be easily done in `R` using the `LDheatmap` package (by default, 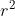 is plotted).
% cat testLD_2.ld | bash ../scripts/LD_blocks.sh chrSIM_21 2000 5000
##### Notes
- LD can be affected by:
- New mutations, genetic drift, population growth (reduces LD)
- Admixture, population structure, inbreeding, natural selection (increases LD)
- 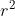 (preferred in popgen)
- ranges between `0` (no linkage) and `1` (markers in complete linkage)
- expected value of `1/2n`
- 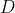 (hard to interpret)
- range depends on frequencies
- sign is arbitrary
- 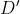
- ranges between `-1` and `1`
- values of `1` or `-1` mean complete linkage
- if frequencies are similar, high values mean the markers are good surrogates for each other
- overestimates LD in small samples
- overestimates LD when one allele is rare
### Hints
* `ngsLD` performance seems to drop considerable under extremely low coverages (<1x); consider these cases only if you have large sample sizes (>100 individuals).
* For some analyses (e.g. LD decay) consider sampling your data (`--rnd_sample`), since `ngsLD` will be much faster and you probably don't need all comparisons.
* For the LD decay, as a rule-of-thumb, consider using at least 10'000 SNPs; check the confidence interval and, if too wide, increase number of SNPs.
### Thread pool
The thread pool implementation was adapted from Mathias Brossard's and is freely available from:
https://github.com/mbrossard/threadpool