https://github.com/kieranrcampbell/clonealign
Bayesian inference of clone-specific gene expression estimates by integrating single-cell RNA-seq and single-cell DNA-seq data
https://github.com/kieranrcampbell/clonealign
cancer-genomics gene-expression single-cell
Last synced: 6 months ago
JSON representation
Bayesian inference of clone-specific gene expression estimates by integrating single-cell RNA-seq and single-cell DNA-seq data
- Host: GitHub
- URL: https://github.com/kieranrcampbell/clonealign
- Owner: kieranrcampbell
- License: apache-2.0
- Created: 2017-11-20T19:43:45.000Z (almost 8 years ago)
- Default Branch: master
- Last Pushed: 2020-12-15T00:51:02.000Z (almost 5 years ago)
- Last Synced: 2024-05-22T18:12:30.946Z (over 1 year ago)
- Topics: cancer-genomics, gene-expression, single-cell
- Language: R
- Homepage:
- Size: 4.11 MB
- Stars: 33
- Watchers: 6
- Forks: 6
- Open Issues: 7
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
- awesome-multi-omics - clonealign - Campbell - gene expression states to clones (scRNA-seq + scDNA-seq (CNV)) - [paper](https://doi.org/10.1101/344309) (Software packages and methods / Single cell multi-omics)
README
# clonealign
[](https://travis-ci.org/kieranrcampbell/clonealign) [](https://zenodo.org/badge/latestdoi/111455172)
`clonealign` assigns single-cell RNA-seq expression to cancer clones by probabilistically mapping RNA-seq to clone-specific copy number profiles using [reparametrization gradient variational inference](https://arxiv.org/abs/1312.6114). This is particularly useful when clones have been inferred using ultra-shallow single-cell DNA-seq meaning SNV analysis is not possible.
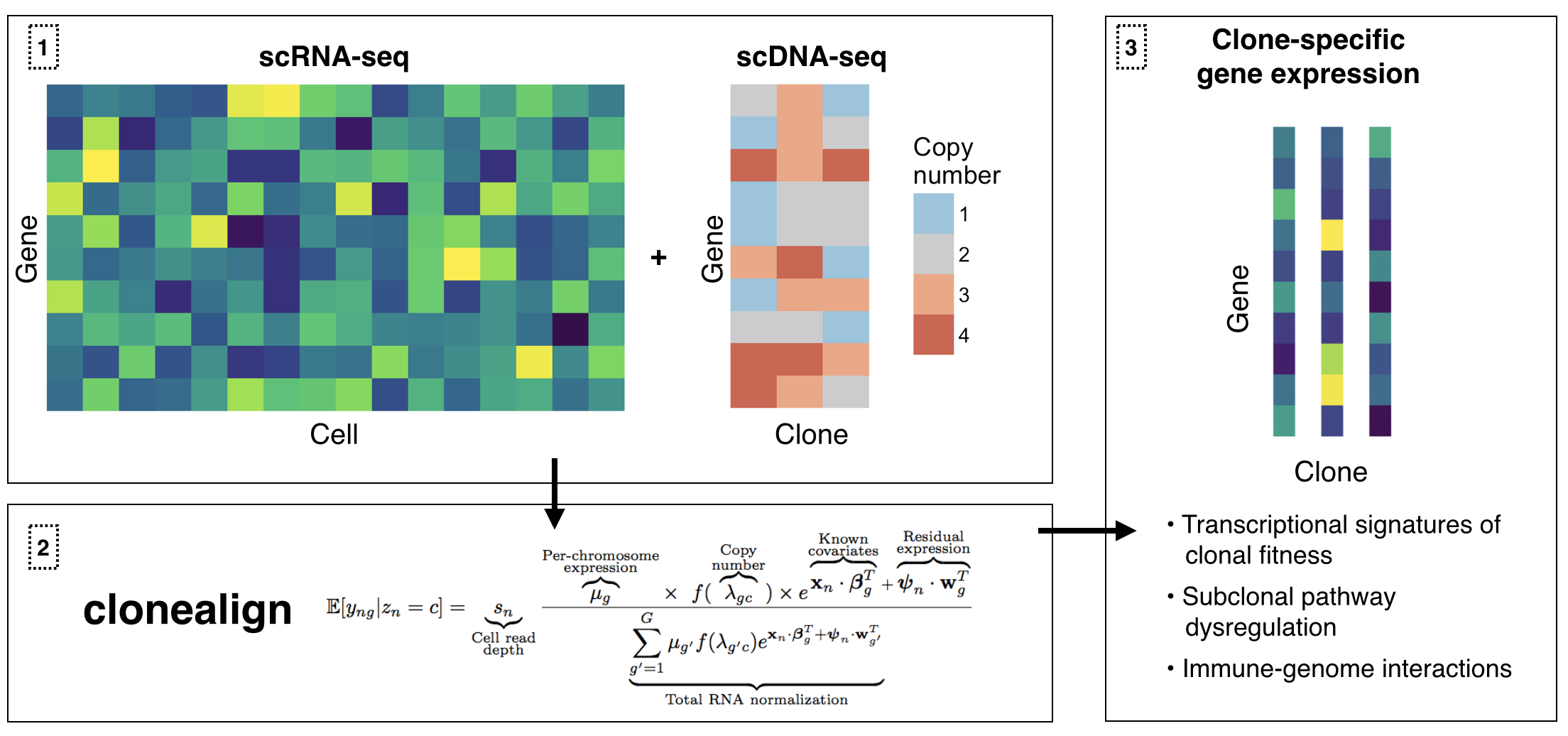
## Version 2.0
Clonealign version 2.0 comes with several updated modelling features. In particular:
* A multinomial likelihood that vastly increases runtime and removes the need for custom size factors
* Multiple restarts through the `run_clonealign` function, where the final fit is chosen as that which
maximizes the ELBO
For more info see the NEWS.md file.
## Getting started
### Vignettes
1. [Introduction to clonealign](https://kieranrcampbell.github.io/clonealign/introduction_to_clonealign.html) Overview of `clonealign` including data preparation, model fitting, plotting results, and advanced inference control
2. [Preparing copy number data for input to clonealign](https://kieranrcampbell.github.io/clonealign/preparing_copy_number_data.html) Instructions for taking region/range specific copy number profiles and converting them to gene and clone specific copy numbers for input to clonealign
### Installation
`clonealign` is built using Google's Tensorflow so requires installation of the R package `tensorflow`. The versioning of Tensorflow and Tensorflow probability currently breaks the standard installation, so the following steps must be taken:
```r
install.packages("tensorflow")
tensorflow::install_tensorflow(extra_packages ="tensorflow-probability", version="2.1.0")
install.packages("devtools") # If not already installed
install_github("kieranrcampbell/clonealign")
```
### Usage
`clonealign` accepts either a cell-by-gene matrix of raw counts or a [SingleCellExperiment](https://bioconductor.org/packages/3.7/bioc/html/SingleCellExperiment.html) with a `counts` assay as gene expression input. It also requires a gene-by-clone matrix or `data.frame` corresponding to the copy number of each gene in each clone. The cells are then assigned to their clones by calling
```r
cal <- clonealign(gene_expression_data, # matrix or SingleCellExperiment
copy_number_data) # matrix or data.frame
print(cal)
```
```
A clonealign_fit for 200 cells, 100 genes, and 3 clones
To access clone assignments, call x$clone
To access ML parameter estimates, call x$ml_params
```
```r
print(head(cal$clone))
```
```
[1] "B" "C" "C" "B" "C" "B"
```
## Paper
[clonealign: statistical integration of independent single-cell RNA and DNA sequencing data from human cancers, _Genome Biology 2019_](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1645-z)
## Authors
Kieran R Campbell, University of British Columbia