https://github.com/lieberinstitute/megadepth
R interface to megadepth: BigWig and BAM related utilities
https://github.com/lieberinstitute/megadepth
bam bigwig daspter megadepth recount2 recount3 rstats
Last synced: about 1 year ago
JSON representation
R interface to megadepth: BigWig and BAM related utilities
- Host: GitHub
- URL: https://github.com/lieberinstitute/megadepth
- Owner: LieberInstitute
- Created: 2020-09-17T13:13:33.000Z (almost 6 years ago)
- Default Branch: devel
- Last Pushed: 2024-12-12T21:45:05.000Z (over 1 year ago)
- Last Synced: 2025-03-31T07:22:10.967Z (over 1 year ago)
- Topics: bam, bigwig, daspter, megadepth, recount2, recount3, rstats
- Language: R
- Homepage: https://lieberinstitute.github.io/megadepth
- Size: 679 KB
- Stars: 12
- Watchers: 5
- Forks: 2
- Open Issues: 6
-
Metadata Files:
- Readme: README.Rmd
- Changelog: NEWS.md
- Contributing: .github/CONTRIBUTING.md
- Code of conduct: .github/CODE_OF_CONDUCT.md
- Support: .github/SUPPORT.md
Awesome Lists containing this project
README
---
output: github_document
---
```{r, include = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
fig.path = "man/figures/README-",
out.width = "100%"
)
```
# megadepth
[](https://lifecycle.r-lib.org/articles/stages.html#stable)
[](https://bioconductor.org/checkResults/release/bioc-LATEST/megadepth)
[](https://bioconductor.org/checkResults/devel/bioc-LATEST/megadepth)
[](http://bioconductor.org/packages/stats/bioc/megadepth/)
[](https://support.bioconductor.org/tag/megadepth)
[](https://bioconductor.org/packages/release/bioc/html/megadepth.html#since)
[](http://bioconductor.org/checkResults/devel/bioc-LATEST/megadepth/)
[](https://bioconductor.org/packages/release/bioc/html/megadepth.html#since)
[](https://codecov.io/gh/LieberInstitute/megadepth?branch=devel)
[](https://github.com/LieberInstitute/megadepth/actions/workflows/check-bioc.yml)
[](https://github.com/LieberInstitute/megadepth/issues)
[](https://github.com/LieberInstitute/megadepth/pulls)
The goal of `megadepth` is to provide an R interface to the command line tool [Megadepth](https://github.com/ChristopherWilks/megadepth) for BigWig and BAM related utilities created by [Christopher Wilks](https://twitter.com/chrisnwilks). This R package enables **fast** processing of BigWig files on downstream packages such as [dasper](https://bioconductor.org/packages/dasper) and [recount3](https://bioconductor.org/packages/recount3). The [Megadepth](https://github.com/ChristopherWilks/megadepth) software also provides utilities for processing BAM files and extracting coverage information from them.
Here is an illustration on how fast `megadepth` is compared to other tools for processing local and remote BigWig files.
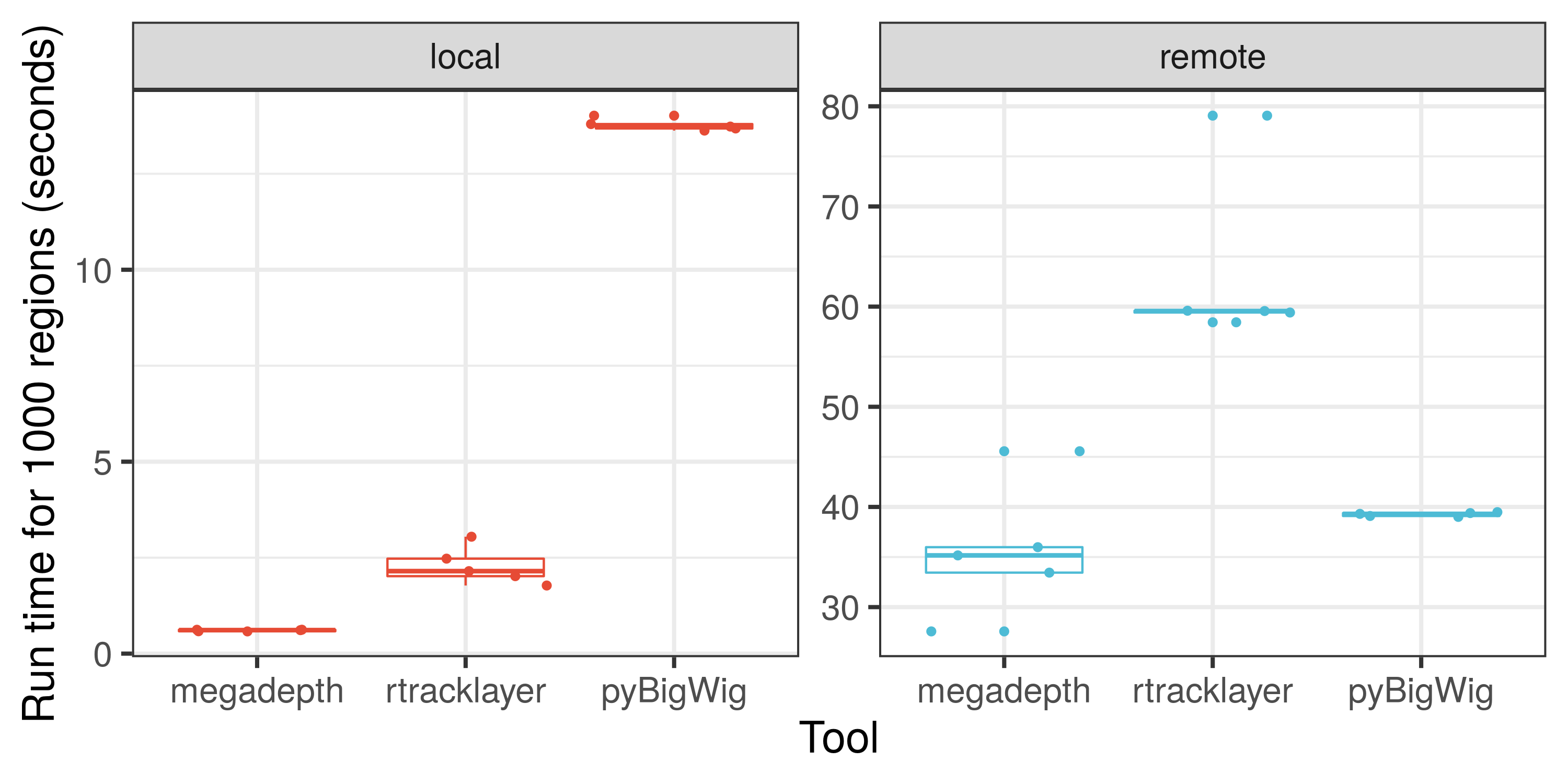
Throughout the documentation we use a capital `M` to refer to the software by Christopher Wilks and a lower case `m` to refer to this R/Bioconductor package.
## Installation instructions
Get the latest stable `R` release from [CRAN](http://cran.r-project.org/). Then install `megadepth` from [Bioconductor](http://bioconductor.org/) using the following code:
```{r 'install', eval = FALSE}
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("megadepth")
```
And the development version from [GitHub](https://github.com/) with:
```{r 'install_dev', eval = FALSE}
BiocManager::install("LieberInstitute/megadepth")
```
## Example
In the following example, we install [Megadepth](https://github.com/ChristopherWilks/megadepth) with `install_megadepth()`, which downloads a binary for your OS (Linux, Windows or macOS). We can then use with an example BigWig file to compute the coverage at a set of regions.
```{r example, eval = requireNamespace('megadepth')}
## Load the R package
library("megadepth")
## Install Megadepth's pre-compiled binary on your system
install_megadepth()
## Next, we locate the example BigWig and annotation files
example_bw <- system.file("tests", "test.bam.all.bw",
package = "megadepth", mustWork = TRUE
)
annotation_file <- system.file("tests", "testbw2.bed",
package = "megadepth", mustWork = TRUE
)
## We can then use megadepth to compute the coverage
bw_cov <- get_coverage(example_bw, op = "mean", annotation = annotation_file)
bw_cov
```
## Full set of utilities
[Megadepth](https://github.com/ChristopherWilks/megadepth) is very powerful and can do a lot of different things. The R/Bioconductor package provides two functions for interfacing with [Megadepth](https://github.com/ChristopherWilks/megadepth), `megadepth_cmd()` and `megadepth_shell()`. For the first one, `megadepth_cmd()`, you need to know the actual command syntax you want to use and format it accordingly. If you are more comfortable with R functions, `megadepth_shell()` uses `r BiocStyle::CRANpkg("cmdfun")` to power this interface and capture the standard output stream into R.
To make it easier to use, `megadepth` includes functions that simplify the number of arguments, read in the output files, and converts them into R/Bioconductor friendly objects, such as `get_coverage()` illustrated above.
We hope that you'll find `megadepth` and [Megadepth](https://github.com/ChristopherWilks/megadepth) useful for your work. If you are interested in checking how **fast** `megadepth` is, check out the [**speed analysis**](https://github.com/LieberInstitute/megadepth/tree/devel/analysis) comparison against other tools. Note that the size of the files used and the number of genomic regions queried will affect the speed comparisons.
```{r "interface_options"}
## R-like interface
## that captures the standard output into R
head(megadepth_shell(help = TRUE))
## Command-like interface
megadepth_cmd("--help")
```
```{r "show_help", echo = FALSE}
x <- megadepth_shell(help = TRUE)
cat(paste0(x, "\n"))
```
## Citation
Below is the citation output from using `citation('megadepth')` in R. Please
run this yourself to check for any updates on how to cite __megadepth__.
```{r 'citation', eval = requireNamespace('megadepth')}
print(citation("megadepth"), bibtex = TRUE)
```
Please note that the `megadepth` was only made possible thanks to many other R and bioinformatics software authors, which are cited either in the vignettes and/or the paper(s) describing this package.
## Code of Conduct
Please note that the `megadepth` project is released with a [Contributor Code of Conduct](https://contributor-covenant.org/version/2/0/CODE_OF_CONDUCT.html). By contributing to this project, you agree to abide by its terms.
## Development tools
* Continuous code testing is possible thanks to [GitHub actions](https://www.tidyverse.org/blog/2020/04/usethis-1-6-0/) through `r BiocStyle::CRANpkg('usethis')`, `r BiocStyle::CRANpkg('remotes')`, `r BiocStyle::Githubpkg('r-hub/sysreqs')` and `r BiocStyle::CRANpkg('rcmdcheck')` customized to use [Bioconductor's docker containers](https://www.bioconductor.org/help/docker/) and `r BiocStyle::Biocpkg('BiocCheck')`.
* Code coverage assessment is possible thanks to [codecov](https://codecov.io/gh) and `r BiocStyle::CRANpkg('covr')`.
* The [documentation website](http://LieberInstitute.github.io/megadepth) is automatically updated thanks to `r BiocStyle::CRANpkg('pkgdown')`.
* The code is styled automatically thanks to `r BiocStyle::CRANpkg('styler')`.
* The documentation is formatted thanks to `r BiocStyle::CRANpkg('devtools')` and `r BiocStyle::CRANpkg('roxygen2')`.
For more details, check the `dev` directory.
This package was developed using `r BiocStyle::Biocpkg('biocthis')`.
## `ReCount` project
The **main documentation website** for all the `recount3`-related projects is available at [**recount.bio**](https://LieberInstitute/github.io/recount3-docs). Please check that website for more information about how this R/Bioconductor package and other tools are related to each other.
## Teams involved
`r BiocStyle::Biocpkg('megadepth')` was made possible to [David Zhang](https://twitter.com/dyzhang32), the author of `r BiocStyle::Biocpkg("dasper")`, and a member of the [Mina Ryten](https://snca.atica.um.es/)'s lab at UCL.
The `ReCount` family involves the following teams:
* [Ben Langmead's lab](http://www.langmead-lab.org/) at JHU Computer Science
* [Kasper Daniel Hansen's lab](https://www.hansenlab.org/) at JHBSPH Biostatistics Department
* [Leonardo Collado-Torres](http://lcolladotor.github.io/) and [Andrew E. Jaffe](http://aejaffe.com/) from [LIBD](https://www.libd.org/)
* [Abhinav Nellore's lab](http://nellore.bio/) at OHSU
* [Jeff Leek's lab](http://jtleek.com/) at JHBSPH Biostatistics Deparment
* Data hosted by [SciServer from IDIES at JHU](https://www.sciserver.org/)
| | | | |
| --- | --- | --- | --- |
|  |
|  |
|  |
|  |
|