https://github.com/mdshw5/fastqp
Simple FASTQ quality assessment using Python
https://github.com/mdshw5/fastqp
bioinformatics fastq kmer-distribution nucleotide-plot python sam
Last synced: 4 months ago
JSON representation
Simple FASTQ quality assessment using Python
- Host: GitHub
- URL: https://github.com/mdshw5/fastqp
- Owner: mdshw5
- License: mit
- Created: 2013-09-23T20:01:47.000Z (almost 13 years ago)
- Default Branch: master
- Last Pushed: 2021-05-22T02:00:12.000Z (about 5 years ago)
- Last Synced: 2025-05-13T17:38:37.317Z (about 1 year ago)
- Topics: bioinformatics, fastq, kmer-distribution, nucleotide-plot, python, sam
- Language: Python
- Homepage: https://pypi.python.org/pypi/fastqp
- Size: 2.57 MB
- Stars: 108
- Watchers: 5
- Forks: 15
- Open Issues: 14
-
Metadata Files:
- Readme: README.md
- License: LICENSE
- Code of conduct: CODE_OF_CONDUCT.md
Awesome Lists containing this project
- Awesome-Bioinformatics - Fastqp - FASTQ and SAM quality control using Python. (Next Generation Sequencing / Sequence Processing)
- awesome-bioinformatics - Fastqp - FASTQ and SAM quality control using Python. (Next Generation Sequencing / Sequence Processing)
README
fastqp
======
[](https://github.com/mdshw5/fastqp/actions/workflows/ci.yml)
[](https://pypi.python.org/pypi/fastqp)
Simple FASTQ, SAM and BAM read quality assessment and plotting using Python.
Features
--------
- Requires only Python with Numpy, Scipy, and Matplotlib libraries
- Works with (gzipped) FASTQ, SAM, and BAM formatted reads
- Tabular, tidy, output statistics so you can create your own graphs
- A useful set of default graphics rivaling comparable QC packages
- Counts *all* IUPAC ambiguous nucleotide codes (NMWSKRYVHDB) if present in sequences
- Downsamples input files to around 2,000,000 reads (user adjustable)
- Allows a 5′ and 3′ (left and right) cycle limit for graphics generation
- Tracks kmers and sequence duplication for the *entire* input file
- Plots base call reference mismatches for aligned reads
- Optional sequence duplication calculation using Bloom filters (beta)
Requirements
------------
Tested on Python 2.7, and 3.4
Tested on Mac OS 10.10 and Linux 2.6.18
Installation
------------
pip install [--user] fastqp
Note: BAM file support requires [samtools](https://github.com/samtools/samtools)
Usage
-----
```
usage: fastqp [-h] [-q] [-s BINSIZE] [-a NAME] [-n NREADS] [-p BASE_PROBS] [-k {2,3,4,5,6,7}] [-o OUTPUT]
[-ll LEFTLIMIT] [-rl RIGHTLIMIT] [-mq MEDIAN_QUAL] [--aligned-only | --unaligned-only] [-d]
input
simple NGS read quality assessment using Python
positional arguments:
input input file (one of .sam, .bam, .fq, or .fastq(.gz) or stdin (-))
optional arguments:
-h, --help show this help message and exit
-q, --quiet do not print any messages (default: False)
-s BINSIZE, --binsize BINSIZE
number of reads to bin for sampling (default: auto)
-a NAME, --name NAME sample name identifier for text and graphics output (default: input file name)
-n NREADS, --nreads NREADS
number of reads sample from input (default: 2000000)
-p BASE_PROBS, --base-probs BASE_PROBS
probabilites for observing A,T,C,G,N in reads (default: 0.25,0.25,0.25,0.25,0.1)
-k {2,3,4,5,6,7}, --kmer {2,3,4,5,6,7}
length of kmer for over-repesented kmer counts (default: 5)
-o OUTPUT, --output OUTPUT
base name for output files (default: fastqp_figures)
-ll LEFTLIMIT, --leftlimit LEFTLIMIT
leftmost cycle limit (default: 1)
-rl RIGHTLIMIT, --rightlimit RIGHTLIMIT
rightmost cycle limit (-1 for none) (default: -1)
-mq MEDIAN_QUAL, --median-qual MEDIAN_QUAL
median quality threshold for failing QC (default: 30)
--aligned-only only aligned reads (default: False)
--unaligned-only only unaligned reads (default: False)
-d, --count-duplicates
calculate sequence duplication rate (default: False)
```
Changes
-------
See [releases page](https://github.com/mdshw5/fastqp/releases) for details.
Examples
--------
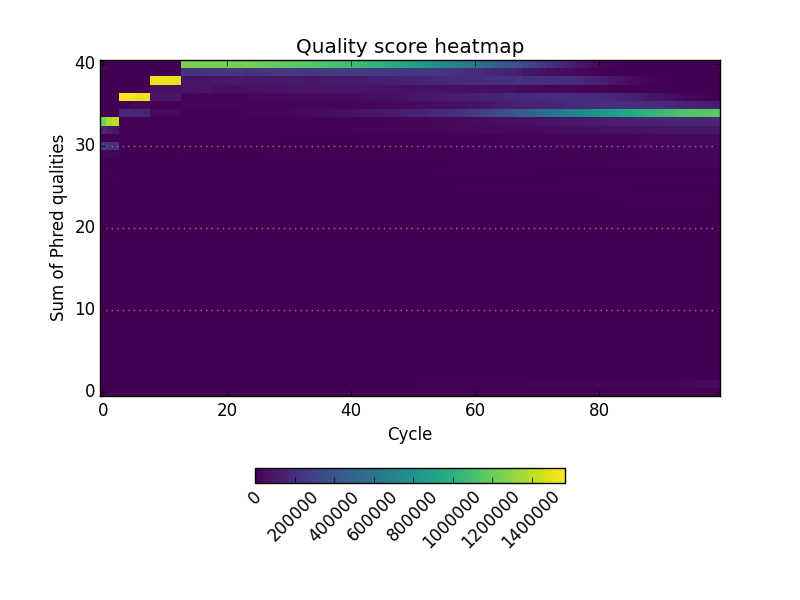
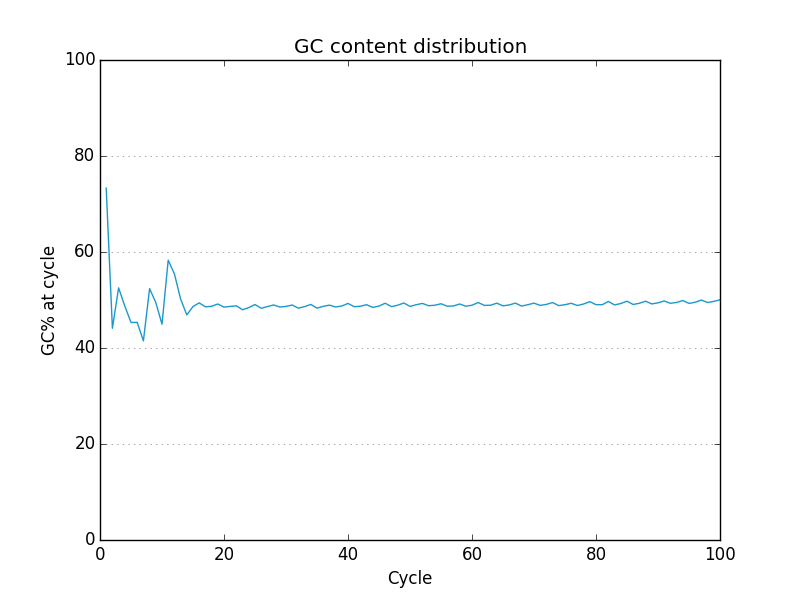
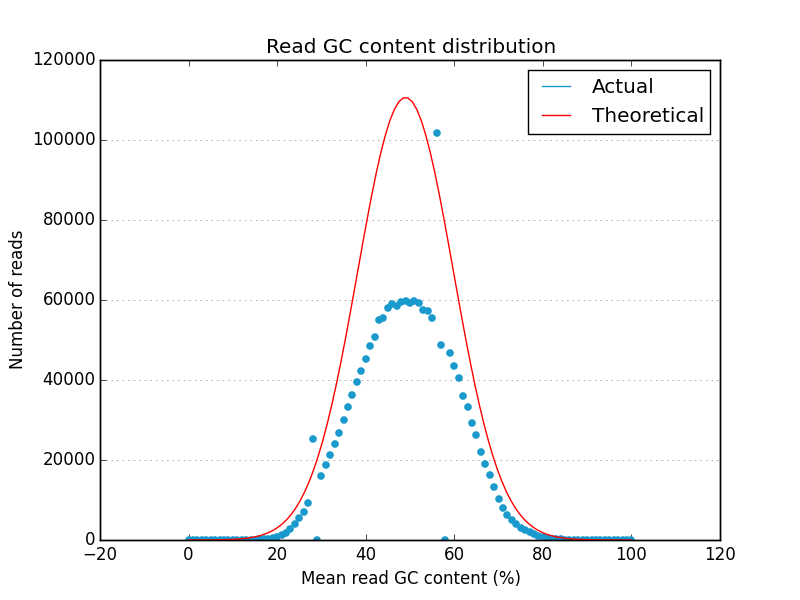
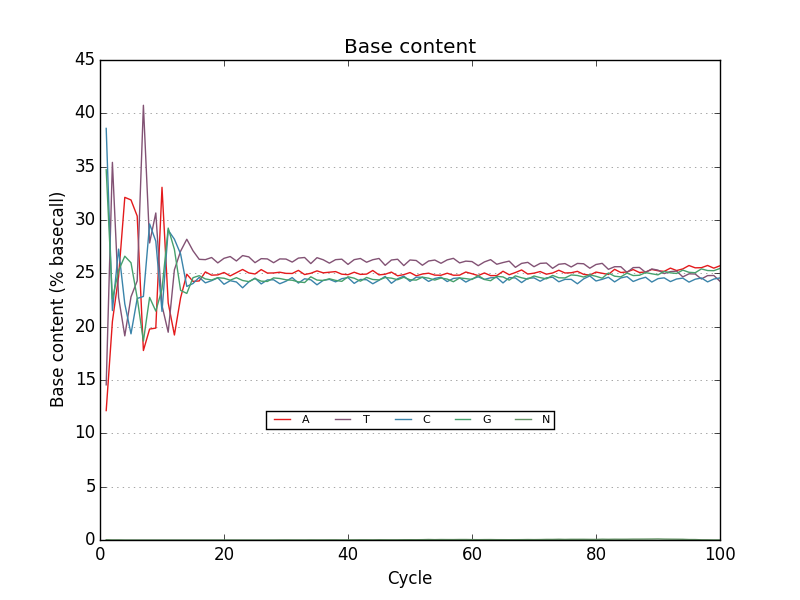
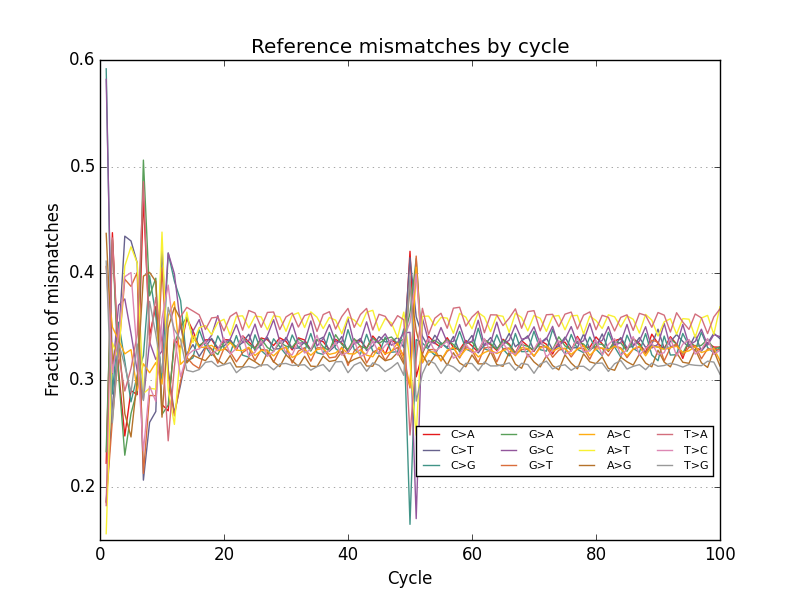
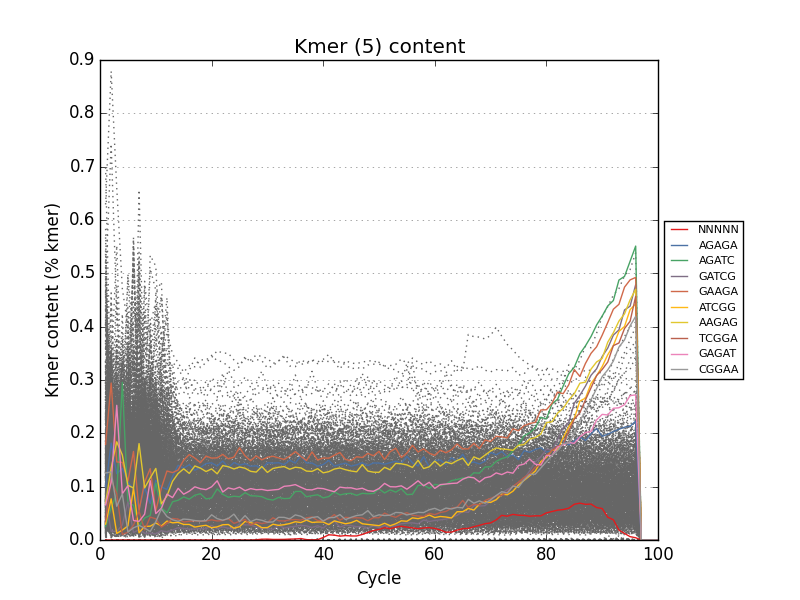
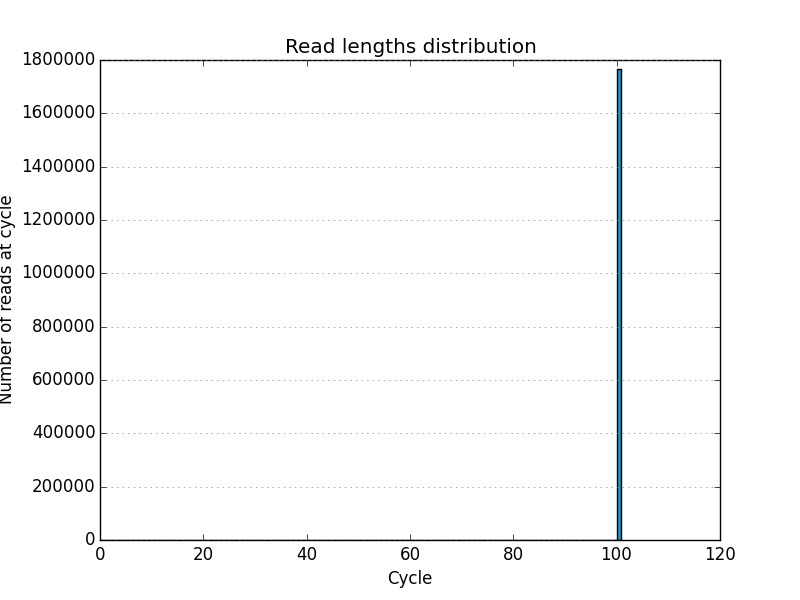
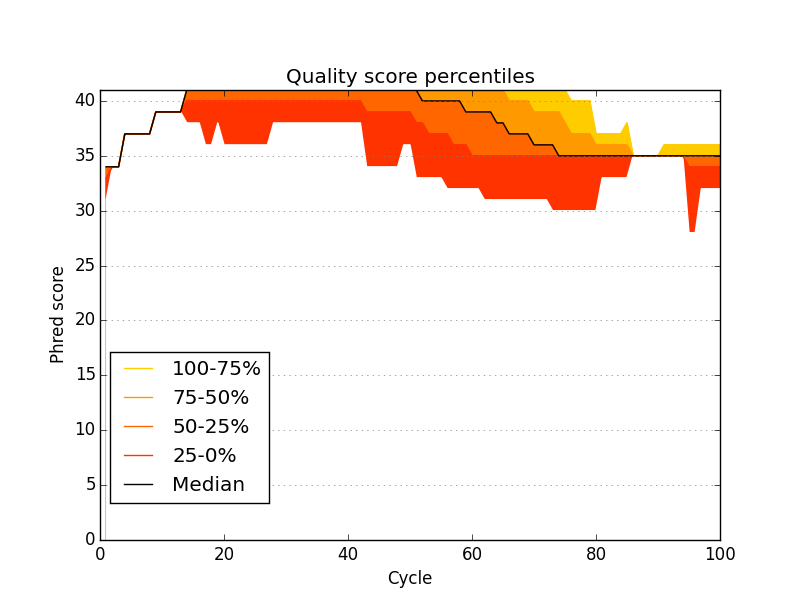
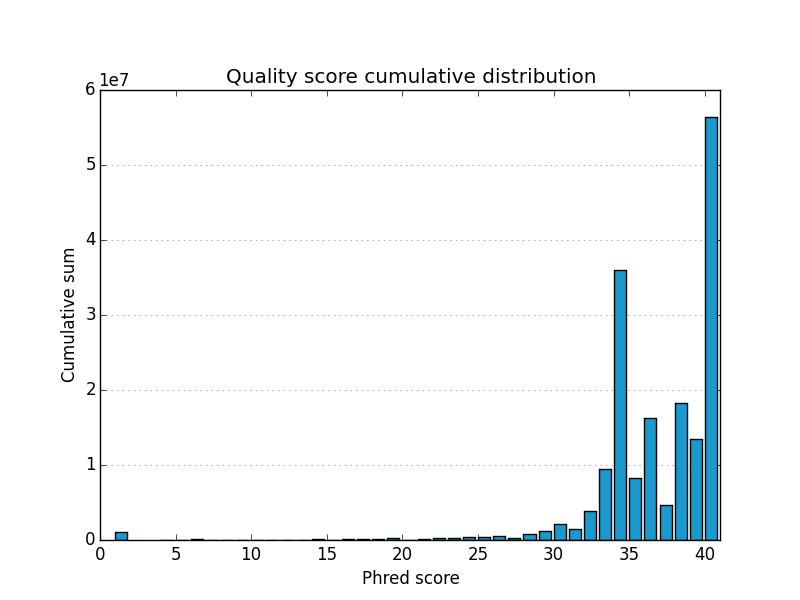
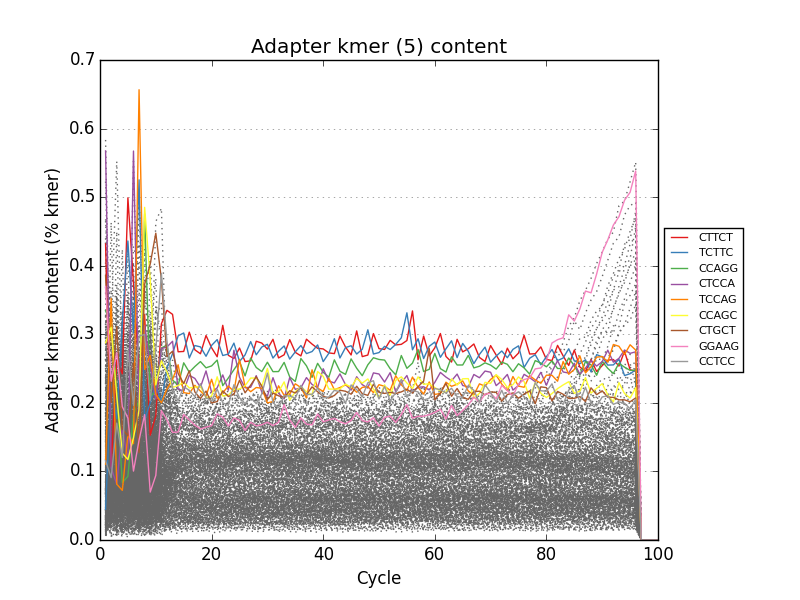
Acknowledgements
----------------
This project is freely licensed by the author, [Matthew Shirley](http://mattshirley.com), and
was completed under the mentorship financial support of Drs. [Sarah Wheelan](http://sjwheelan.som.jhmi.edu)
and [Vasan Yegnasubramanian](http://yegnalab.onc.jhmi.edu) at the Sidney Kimmel Comprehensive
Cancer Center in the Department of Oncology.