https://github.com/membrizard/ml_conformer_generator
Shape-constrained molecule generation via Equivariant Diffusion and GCN
https://github.com/membrizard/ml_conformer_generator
chemistry conformers diffusion-models graph-convolutional-networks molecule-generation rdkit
Last synced: 3 months ago
JSON representation
Shape-constrained molecule generation via Equivariant Diffusion and GCN
- Host: GitHub
- URL: https://github.com/membrizard/ml_conformer_generator
- Owner: Membrizard
- License: apache-2.0
- Created: 2025-01-21T10:06:01.000Z (over 1 year ago)
- Default Branch: main
- Last Pushed: 2025-08-09T15:27:50.000Z (11 months ago)
- Last Synced: 2025-08-09T17:28:13.171Z (11 months ago)
- Topics: chemistry, conformers, diffusion-models, graph-convolutional-networks, molecule-generation, rdkit
- Language: Python
- Homepage:
- Size: 34.7 MB
- Stars: 11
- Watchers: 3
- Forks: 1
- Open Issues: 0
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
README
# ML Conformer Generator
[](https://doi.org/10.1039/D5DD00318K)
**ML Conformer Generator**
is a tool for spatially-aware molecule generation with an Equivariant Diffusion Model (EDM)
and a Graph Convolutional Network (GCN). It is designed to generate 3D molecular conformations
that are both chemically valid and spatially similar to a reference shape.
---
## Molecule Generation in Action
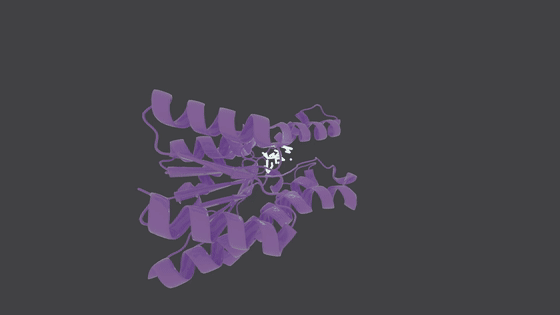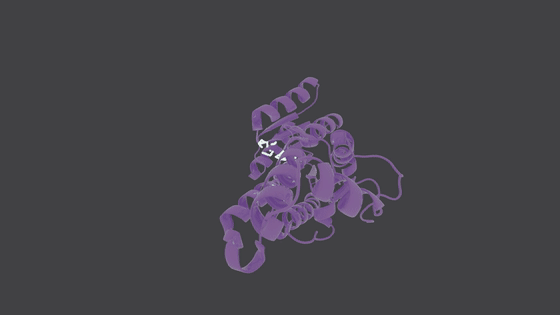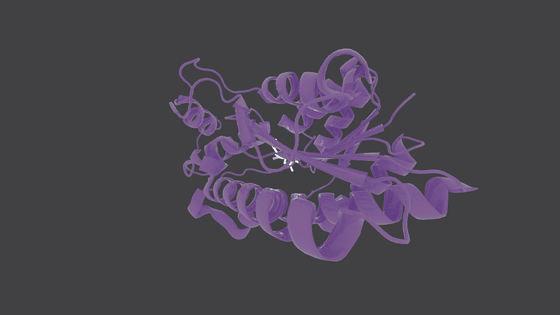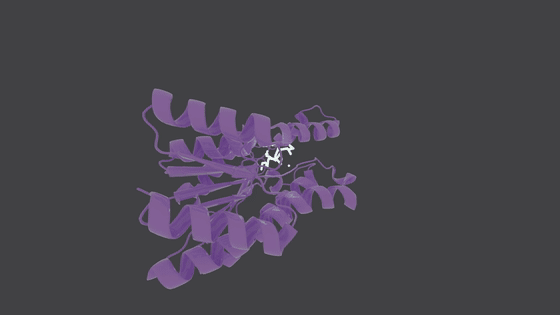

---
## Supported features
* **Shape-guided molecular generation**
Generate novel molecules that conform to arbitrary 3D shapes—such as protein binding pockets or custom-defined spatial regions.
* **Objective-guided Generation**
Use reinforcement learning (RL) to steer molecular generation toward higher-scoring candidates, with support for custom scoring functions.
* **Reference-based conformer similarity**
Create molecules conformations of which closely resemble a reference structure, supporting scaffold-hopping and ligand-based design workflows.
* **Fragment-based inpainting**
Fix specific substructures or fragments within a molecule and complete or grow the rest in a geometrically consistent manner.
* **Inertial Fragment Matching**
Generate molecules fragment-by-fragment by leveraging the physical properties of the shape descriptor, improving both shape similarity and chemical validity.
## Citation
If you use **MLConfGen** in your research, please cite:
Denis Sapegin, Fedor Bakharev, Dmitry Krupenya, Azamat Gafurov, Konstantin Pildish, and Joseph C. Bear.
*Moment of inertia as a simple shape descriptor for diffusion-based shape-constrained molecular generation.*
Digital Discovery, 2025.
DOI: [10.1039/D5DD00318K](https://doi.org/10.1039/D5DD00318K)
---
## Installation
1. Install the package for your preferred backend:
* `pip install mlconfgen[torch]` — use the PyTorch-based inference pipeline
* `pip install mlconfgen[onnx]` — use the torch-free ONNX runtime version
2. Load the weights from Huggingface
> https://huggingface.co/Membrizard/ml_conformer_generator
`edm_moi_chembl_15_39.pt`
`adj_mat_seer_chembl_15_39.pt`
---
## 🐍 Python API
See interactive examples: `./python_api_demo.ipynb`
```python
from rdkit import Chem
from mlconfgen import MLConformerGenerator, evaluate_samples
model = MLConformerGenerator(
edm_weights="./edm_moi_chembl_15_39.pt",
adj_mat_seer_weights="./adj_mat_seer_chembl_15_39.pt",
diffusion_steps=100,
)
reference = Chem.MolFromMolFile('./assets/demo_files/ceyyag.mol')
samples = model.generate_conformers(reference_conformer=reference, n_samples=20, variance=2)
aligned_reference, std_samples = evaluate_samples(reference, samples)
```
---
## 🚀 Overview
This solution employs:
- **Equivariant Diffusion Model (EDM) [[1]](https://doi.org/10.48550/arXiv.2203.17003)**: For generating atom coordinates and types under a shape constraint.
- **Graph Convolutional Network (GCN) [[2]](https://doi.org/10.1039/D3DD00178D)**: For predicting atom adjacency matrices.
- **Deterministic Standardization Pipeline**: For refining and validating generated molecules.
---
## 🧠 Model Training
- Trained on **1.6 million** compounds from the **ChEMBL** database.
- Filtered to molecules with **15–39 heavy atoms**.
- Supported elements: `H, C, N, O, F, P, S, Cl, Br`.
---
## 🧪 Standardization Pipeline
The generated molecules are post-processed through the following steps:
- Largest Fragment picker
- Valence check
- Kekulization
- RDKit sanitization
- Constrained Geometry optimization via **MMFF94** Molecular Dynamics
---
## 📏 Evaluation Pipeline
Aligns and Evaluates shape similarity between generated molecules and a reference using
**Shape Tanimoto Similarity [[3]](https://doi.org/10.1007/978-94-017-1120-3_5 )** via Gaussian Molecular Volume overlap.
> Hydrogens are ignored in both reference and generated samples for this metric.
---
## 📊 Performance (100 Denoising Steps)
*Tested on 100,000 samples using 1,000 CCDC Virtual Screening [[4]](https://www.ccdc.cam.ac.uk/support-and-resources/downloads/) reference compounds.*
### General Overview
- ⏱ **Avg time to generate 50 valid samples**: 11.46 sec (NVIDIA H100) (100 samples batch)
- ⚡️ **Generation speed**: 4.18 valid molecules/sec (100 samples batch)
- 💾 **GPU memory (per generation thread)**: Up to 14.0 GB (`float16` 39 atoms 100 samples)
- 📐 **Avg Shape Tanimoto Similarity**: 53.32% (Basic generation) - 69.97% (Inertial Fragment Matching)
- 🎯 **Max Shape Tanimoto Similarity**: 99.69%
- 🔬 **Avg Chemical Tanimoto Similarity (2-hop 2048-bit Morgan Fingerprints)**: 10.87%
- 🧬 **% Chemically novel (vs. training set)**: 99.84%
- ✔️ **% Valid molecules (post-standardization)**: 48% (ML Bond Prediction) - 93% (OpenBabel bond prediction)
- 🔁 **% Unique molecules in generated set**: 99.94%
- 📎 **Fréchet Fingerprint Distance (2-hop 2048-bit Morgan Fingerprints)**:
- To ChEMBL: 4.13
- To PubChem: 2.64
- To ZINC (250k): 4.95
### PoseBusters [[5]](https://doi.org/10.1039/D3SC04185A) validity check results:
**Overall stats**:
- PB-valid molecules: **91.33 %**
**Detailed Problems**:
- position: 0.01 %
- mol_pred_loaded: 0.0 %
- sanitization: 0.01 %
- inchi_convertible: 0.01 %
- all_atoms_connected: 0.0 %
- bond_lengths: 0.24 %
- bond_angles: 0.70 %
- internal_steric_clash: 2.31 %
- aromatic_ring_flatness: 3.34 %
- non-aromatic_ring_non-flatness: 0.27 %
### Synthesizability of the generated compounds
#### SA Score [[6]](https://doi.org/10.1186/1758-2946-1-8)
*1 (easy to make) - 10 (very difficult to make)*
**Average SA Score**: **3.18**
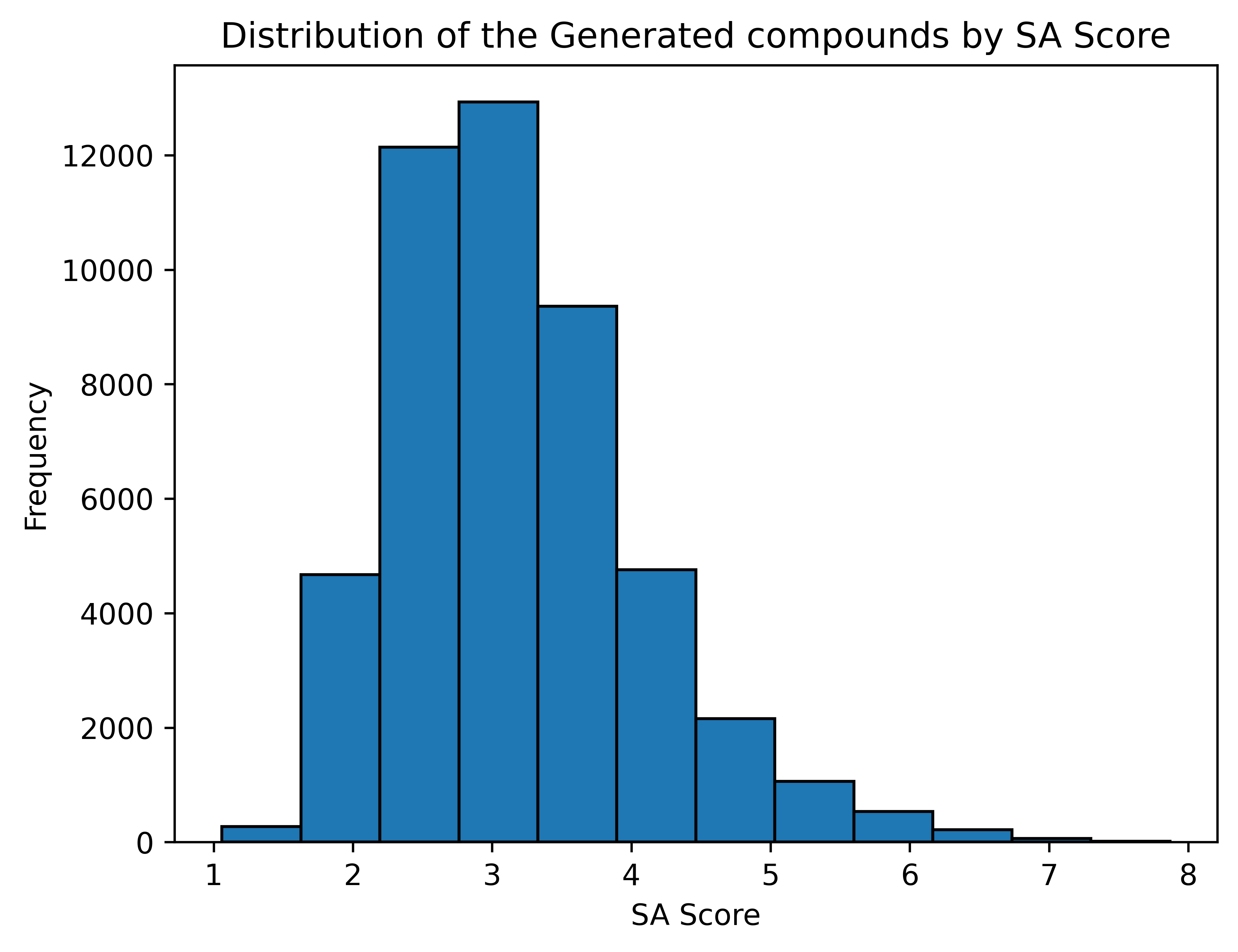
---
## RL Fine Tuning
MLConformerGenerator supports objective-guided reinforcement learning (RL) fine-tuning, allowing you to steer the generated molecular distribution toward molecules that better match your desired properties.
Scoring functions are fully customizable. The only requirement is that they accept a list of RDKit `Mol` objects and return a list of scores in the range `[0, 1]`.
A scoring function should follow this interface:
```python
from rdkit import Chem
def scoring_function(mols: list[Chem.Mol | None]) -> list[float]:
...
```
### Example: RL fine-tuning
> [!NOTE]
> If `scoring_function` is None, a default scoring function enforcing validity is applied for RL.
```python
from rdkit import Chem
from mlconfgen import MLConformerGenerator
model = MLConformerGenerator(
edm_weights="./edm_moi_chembl_15_39.pt",
adj_mat_seer_weights="./adj_mat_seer_chembl_15_39.pt",
diffusion_steps=10,
)
reference = Chem.MolFromMolFile('./assets/demo_files/ceyyag.mol')
model.fine_tune(
reference_conformer=reference,
variance=1,
n_epochs=20,
sigma=60.0,
lambda_edm_adapter=1.5,
temperature=1.5,
n_samples_per_mol=16,
eval_every=5,
save_dir="./rl_checkpoints"
)
```
Fine-tuning produces both the best and the latest checkpoints, which can later be loaded into the model:
```python
from mlconfgen import MLConformerGenerator
model = MLConformerGenerator(
edm_weights="./edm_moi_chembl_15_39.pt",
adj_mat_seer_weights="./adj_mat_seer_chembl_15_39.pt",
finetune_checkpoint = "./finetune_checkpoint.pt",
diffusion_steps=10,
)
# Or
model.load_finetune_checkpoint("./finetune_checkpoint.pt")
```
### REINVENT4 compatibility
The RL fine-tuning pipeline is compatible with scoring functions from [REINVENT4](https://github.com/MolecularAI/REINVENT4/tree/main).
If REINVENT4 is installed, you can use `ReinventScoreWrapper` to load a REINVENT4 scoring configuration and use MLConfGen as a spatially-aware molecule generator.
For working examples, see `rl_fine_tuning_demo.ipynb.`
```python
from rdkit import Chem
from mlconfgen import MLConformerGenerator
from mlconfgen.rl_fine_tuning.reinvent_score_wrapper import ReinventScoreWrapper
model = MLConformerGenerator(
edm_weights="./edm_moi_chembl_15_39.pt",
adj_mat_seer_weights="./adj_mat_seer_chembl_15_39.pt",
diffusion_steps=10,
)
reference = Chem.MolFromMolFile('./assets/demo_files/ceyyag.mol')
scoring_function = ReinventScoreWrapper("./assets/demo_files/scoring_config.toml")
model.fine_tune(
scoring_function=scoring_function,
reference_conformer=reference,
variance=1,
n_epochs=100,
train_batch_size=128,
eval_batch_size=128,
learning_rate= 8e-5,
sigma=128.0,
lambda_edm_adapter=1.5,
lambda_edm_reg=0.2,
temperature=1.5,
n_samples_per_mol=32,
eval_every=5,
save_dir="./rl_checkpoints_reinvent",
)
```
---
## Generation Examples
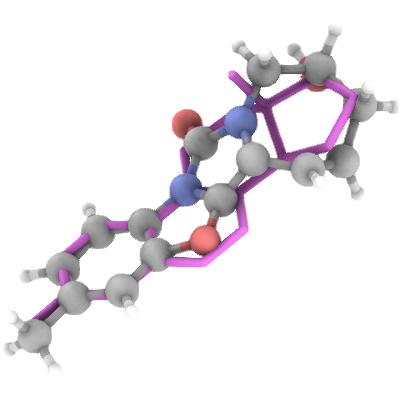
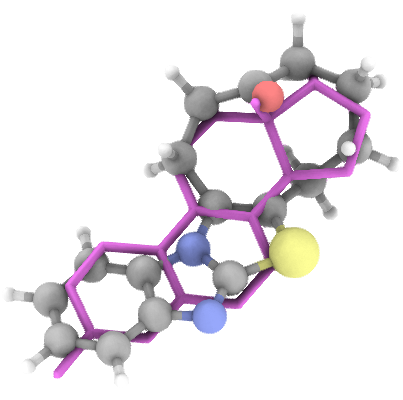
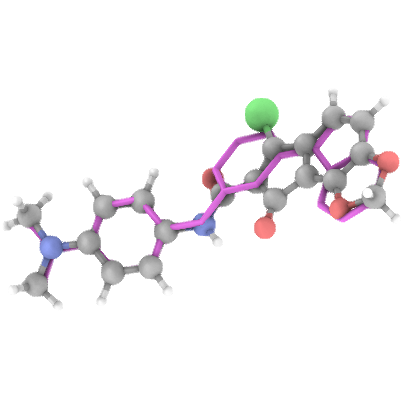
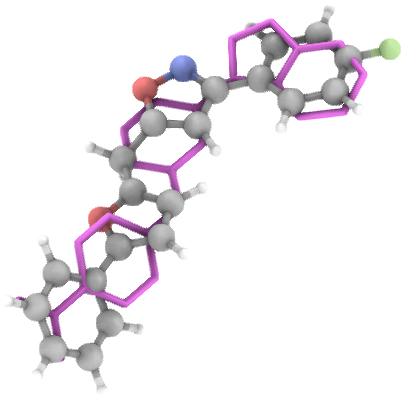
---
## 💾 Access & Licensing
The **Python package and inference code are available on GitHub** under Apache 2.0 License
> https://github.com/Membrizard/ml_conformer_generator
The trained model **Weights** are available at
> https://huggingface.co/Membrizard/ml_conformer_generator
And are licensed under CC BY-NC-ND 4.0
The usage of the trained weights for any profit-generating activity is restricted.
For commercial licensing and inference-as-a-service, contact:
[Denis Sapegin](https://github.com/Membrizard)
---
## ONNX Inference:
For torch Free inference an ONNX version of the model is present.
Weights of the model in ONNX format are available at:
> https://huggingface.co/Membrizard/ml_conformer_generator
`egnn_chembl_15_39.onnx`
`adj_mat_seer_chembl_15_39.onnx`
```python
from mlconfgen import MLConformerGeneratorONNX
from rdkit import Chem
model = MLConformerGeneratorONNX(
egnn_onnx="./egnn_chembl_15_39.onnx",
adj_mat_seer_onnx="./adj_mat_seer_chembl_15_39.onnx",
diffusion_steps=100,
)
reference = Chem.MolFromMolFile('./assets/demo_files/yibfeu.mol')
samples = model.generate_conformers(reference_conformer=reference, n_samples=20, variance=2)
```
Install ONNX GPU runtime (if needed):
`pip install onnxruntime-gpu`
---
## Export to ONNX
An option to compile the model to ONNX is provided
requires `onnxscript==0.2.2`
`pip install onnxscript`
```python
from mlconfgen import MLConformerGenerator
from onnx_export import export_to_onnx
model = MLConformerGenerator()
export_to_onnx(model)
```
This compiles and saves the ONNX files to: `./`
---
## Testing
To execute all tests (including slow generation ones)
`pytest -v tests`
To bypass generation tests
`pytest -v tests -m "not slow"`
---
## Streamlit App
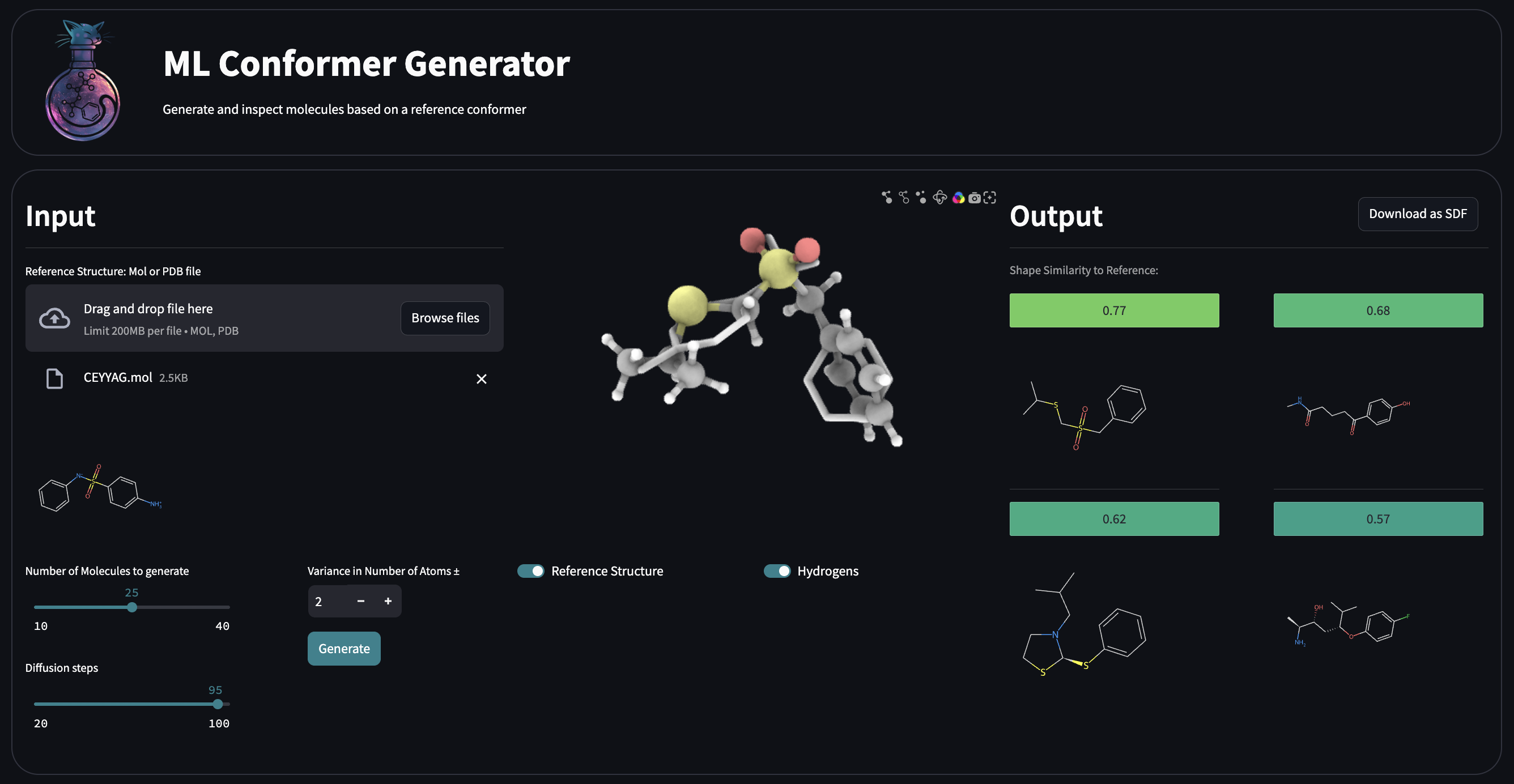
### Running
- Move the trained PyTorch weights into `./streamlit_app`
`./streamlit_app/edm_moi_chembl_15_39.pt`
`./streamlit_app/adj_mat_seer_chembl_15_39.pt`
- Install the dependencies `pip install -r ./streamlit_app/requirements.txt`
- Bring the app UI up:
```commandline
cd ./streamlit_app
streamlit run app.py
```
### Streamlit App Development
1. To enable development mode for the 3D viewer (`stspeck`), set `_RELEASE = False` in `./streamlit/stspeck/__init__.py`.
2. Navigate to the 3D viewer frontend and start the development server:
```commandline
cd ./frontend/speck/frontend
npm run start
```
This will launch the dev server at `http://localhost:3001`
3. In a separate terminal, run the Streamlit app from the root frontend directory:
```commandline
cd ./streamlit_app
streamlit run app.py
```
4. To build the production version of the 3D viewer, run:
```commandline
cd ./streamlit_app/stspeck/frontend
npm run build
```