https://github.com/mlandis/phyddle
Phylogenetic model exploration with deep learning
https://github.com/mlandis/phyddle
deep-learning epidemiology evolution phylogenetics statistical-models stochastic-processes
Last synced: 6 months ago
JSON representation
Phylogenetic model exploration with deep learning
- Host: GitHub
- URL: https://github.com/mlandis/phyddle
- Owner: mlandis
- License: other
- Created: 2023-03-17T01:45:37.000Z (over 3 years ago)
- Default Branch: main
- Last Pushed: 2026-01-18T23:17:41.000Z (6 months ago)
- Last Synced: 2026-01-19T08:45:23.943Z (6 months ago)
- Topics: deep-learning, epidemiology, evolution, phylogenetics, statistical-models, stochastic-processes
- Language: Python
- Homepage: https://phyddle.org
- Size: 267 MB
- Stars: 8
- Watchers: 4
- Forks: 0
- Open Issues: 1
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
README
# phyddle 0.3.0
Software for exploring phylogenetic models with deep learning [[manuscript](https://doi.org/10.1093/sysbio/syaf036)]
## User guide
Visit https://phyddle.org to learn how to use the software.
## Overview
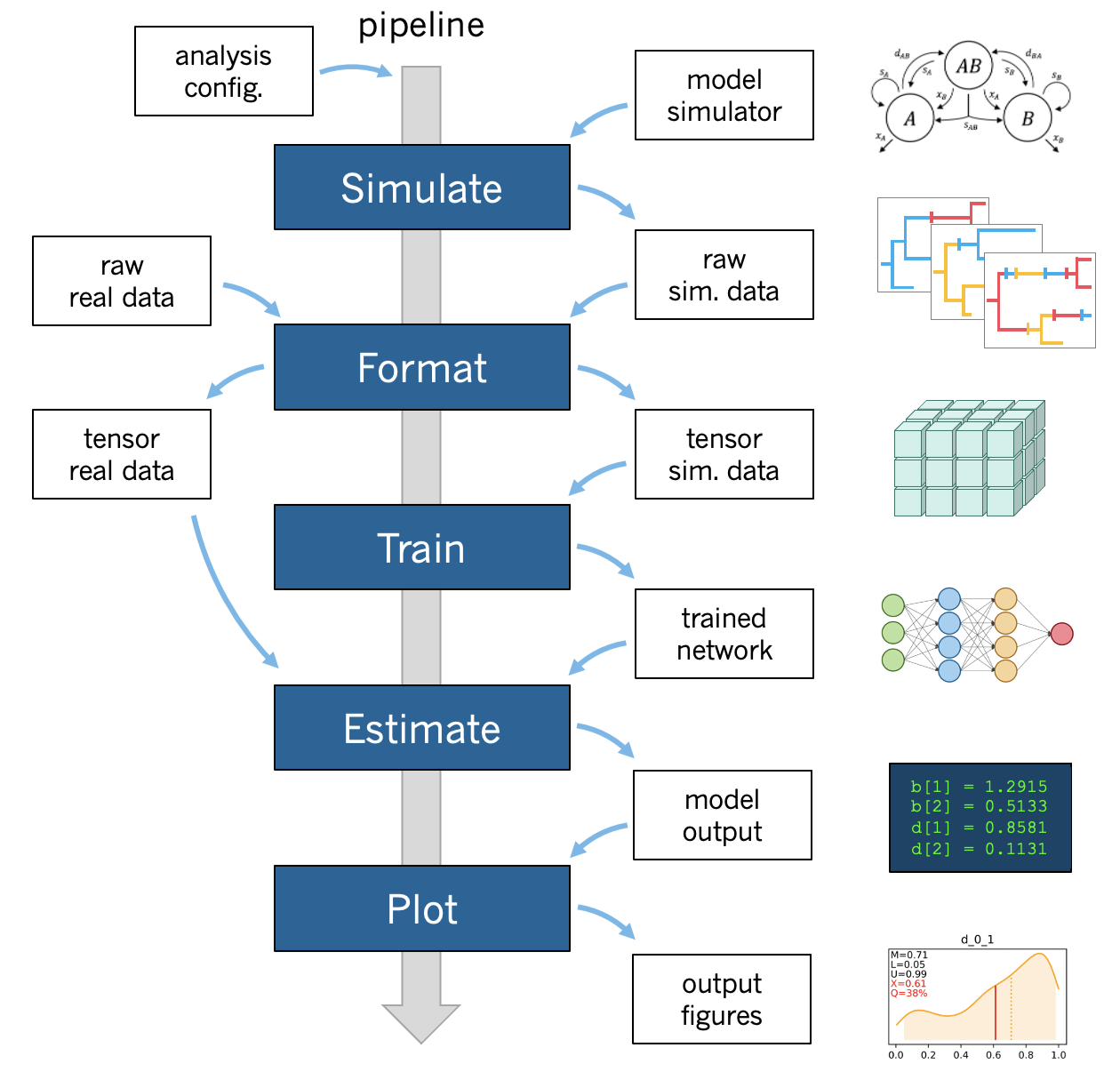
A standard phyddle analysis performs the following tasks for you:
- **Pipeline configuration** applies analysis settings provided through a config file and/or command line arguments.
- **Simulate** simulates a large training dataset using a user-designed simulator.
- **Format** encodes the raw simulated data (from *Simulate*) into tensor format for *Train*.
- **Train** loads and splits training data (from *Format*), builds a network, then trains and saves the network.
- **Estimate** estimates model parameters for a new dataset with the trained network (from *Train*).
- **Plot** generates figures that summarize the training data (*Format*), the network and its training (*Train*), and any new predictions (*Estimate*).
## Quick start
To run a phyddle analysis enter the `scripts` directory:
```shell
cd ~/projects/phyddle
```
Then create and run a pipeline under the settings you've specified in `workspace/example/config.py`:
```shell
cd workspace/example
phyddle --cfg config.py
```
This will run a phyddle analysis with 1000 simulations using R and the castor package for a simple birth-death model with one 3-state character. In practice, you'll want to generate a larger training dataset with anywhere from 10k to 1M examples, depending on the model.
To add new examples to your training set
```shell
# simulate new training examples and store in simulate
phyddle -s S -c config.py --sim_more 14000
# encode all raw_data examples as tensors in format
phyddle -s F -c config.py
# train network with tensor data, but override batch size, then store in train
phyddle -s T -c config.py --trn_batch_size 256
# make prediction for empirical example in dataset
phyddle -s E -c config.py
# generate figures and store in plot
phyddle -s P -c config.py
```
To see a full list of all options currently supported by phyddle
```shell
phyddle --help
```
## Installation
A stable version of phyddle can be installed using the Python package manager, pip:
```shell
python3 -m pip install --upgrade phyddle
# ... install ...
phyddle
```
...or using conda:
```shell
conda create -n phyddle_env -c bioconda -c landismj phyddle
# ... install ...
conda activate phyddle_env
phyddle
```
phyddle uses third-party simulators to generate training datasets. Example workflows assume that [R](https://cran.r-project.org), [RevBayes](https://revbayes.github.io), [Phylojunction](https://phylojunction.org/build/html/index.html), or [BEAST](https://www.beast2.org/) with [MASTER](https://github.com/tgvaughan/MASTER) (plugin) is installed on your machine and can be executed as a command from terminal. The documentation explains how to configure R for use with phyddle.
## Need help?
Visit the [Discussions](https://github.com/mlandis/phyddle/discussions) page to interact with other phyddle users and receive help.
## Citation
If you used phyddle, please cite:
> MJ Landis, A Thompson. 2025. phyddle: software for exploring phylogenetic models with deep learning. Systematic Biology (in press). doi:10.1093/sysbio/syaf036.
> A Thompson, B Liebeskind, EJ Scully, MJ Landis. 2024. Deep learning and likelihood approaches for viral phylogeography converge on the same answers whether the inference model is right or wrong. Systematic Biology 73:183-206.
## Note on code stability
Code on the [main](https://github.com/mlandis/phyddle/tree/main) branch is tested and stable with respect to the standard use cases. Code on the [development](https://github.com/mlandis/phyddle/tree/development) branch contains new features, but is not as rigorously tested. Most phyddle development occurs on a 16-core Intel Macbook Pro laptop and a 64-core Intel Ubuntu server. Any feedback is appreciated! [michael.landis@wustl.edu](mailto:michael.landis@wustl.edu)
## About
Thanks for your interest in phyddle. The phyddle project emerged from a phylogenetic deep learning study led by Ammon Thompson ([paper](https://doi.org/10.1093/sysbio/syad074)). The goal of phyddle is to provide its users with a generalizable pipeline workflow for phylogenetic modeling and deep learning. This hopefully will make it easier for phylogenetic model enthusiasts and developers to explore and apply models that do not have tractable likelihood functions. It's also intended for use by methods developers who want to characterize how deep learning methods perform under different conditions for standard phylogenetic estimation tasks. Read more about phyddle at https://doi.org/10.1093/sysbio/syaf036.
The phyddle project is developed by [Michael Landis](https://landislab.org) and [Ammon Thompson](https://scholar.google.com/citatio:wqns?user=_EpmmTwAAAAJ&hl=en&oi=ao).