https://github.com/plger/scdblfinder
Methods for detecting doublets in single-cell sequencing data
https://github.com/plger/scdblfinder
doublets single-cell
Last synced: about 1 year ago
JSON representation
Methods for detecting doublets in single-cell sequencing data
- Host: GitHub
- URL: https://github.com/plger/scdblfinder
- Owner: plger
- License: gpl-3.0
- Created: 2019-05-05T13:03:47.000Z (about 7 years ago)
- Default Branch: devel
- Last Pushed: 2024-03-20T10:38:40.000Z (about 2 years ago)
- Last Synced: 2024-04-20T15:00:29.087Z (about 2 years ago)
- Topics: doublets, single-cell
- Language: R
- Homepage: https://plger.github.io/scDblFinder/
- Size: 12.3 MB
- Stars: 130
- Watchers: 5
- Forks: 17
- Open Issues: 4
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
README
# scDblFinder 
[](https://github.com/plger/scDblFinder/actions)
The `scDblFinder` package gathers various methods for the detection and handling of doublets/multiplets in single-cell sequencing data (i.e. multiple cells captured within the same droplet or reaction volume), including the novel `scDblFinder` method.
The methods included here are _complementary_ to doublets detection via cell hashes and SNPs in multiplexed samples: while hashing/genotypes can identify doublets formed by cells of the same type (homotypic doublets) from two samples, which are often nearly undistinguishable from real cells transcriptionally (and hence generally unidentifiable through the present package), it cannot identify doublets made by cells of the same sample, even if they are heterotypic (formed by different cell types). Instead, the methods presented here are primarily geared towards the identification of heterotypic doublets, which for most purposes are also the most critical ones.
For a brief overview of the methods, see the [introductory vignette](https://plger.github.io/scDblFinder/articles/introduction.html) (`vignette("introduction", package="scDblFinder")`). For the detailed study including comparison with alternative methods, see the [paper](https://doi.org/10.12688/f1000research.73600.2). Here, we will showcase doublet detection using the fast and comprehensive `scDblFinder` method.
***Important update*: the scDblFinder version (1.20) initially shipped with Bioconductor 3.20 (current) had a wrong default doublet rate argument. This has been fixed in Bioconductor, but you should update your package.**
## Getting started
### Installation
You may install the pakage using:
```r
BiocManager::install("scDblFinder")
```
Or, to get the very latest version,
```r
BiocManager::install("plger/scDblFinder")
```
The latest version will not be compatible with older Bioconductor versions.
Note that, when not installing from git, Bioconductor does not install the latest version of packages, but (to ensure compatibility between packages) installs the version tied to your Bioconductor version. To ensure the best results, install the latest Bioconductor release. We recommend to avoid using scDblFinder from versions prior to Bioconductor 3.14, which give suboptimal results, and scATAC users will need scDblFinder version 1.13.2 or above.
Finally, the documentation here refers to the latest version. If you are using an earlier Bioconductor release, the more accurate documentation will be that of your version, available either from bioconductor or from `vignette("introduction", package="scDblFinder")`.
### Basic usage
Given an object `sce` of class `SingleCellExperiment` (which does not contain any empty drops, but hasn't been further filtered), you can launch the doublet detection with:
```r
library(scDblFinder)
sce <- scDblFinder(sce)
```
This will add a number of columns to the `colData` of `sce`, the most important of which are:
* `sce$scDblFinder.score` : the final doublet score (the higher the more likely that the cell is a doublet)
* `sce$scDblFinder.class` : the classification (doublet or singlet)
There are several additional columns containing further information (e.g. the most likely origin of the putative doublet), an overview of which is available in the [vignette](https://plger.github.io/scDblFinder/articles/scDblFinder.html) (`vignette("scDblFinder")`).
### Multiple samples
If you have multiple samples (understood as different cell captures, i.e. for multiplexed samples with cell hashes, rather use the batch), then it is preferable to provide `scDblFinder` with this information in order to take into consideration batch/sample-specific doublet rates. You can do this by simply providing a vector of the sample ids to the `samples` parameter of scDblFinder or, if these are stored in a column of `colData`, the name of the column. With default settings, the this will result in samples being processed separately, which appears to be faster, more robust to batch effects, and as accurate as training a single model (see the `multiSampleMode` argument for other options).
In such cases, you might also consider multithreading it using the `BPPARAM` parameter. For example:
```r
library(BiocParallel)
sce <- scDblFinder(sce, samples="sample_id", BPPARAM=MulticoreParam(3))
table(sce$scDblFinder.class)
```
### Cluster-based detection
`scDblFinder` has two main modes for generating artificial doublets: a random one (`clusters=FALSE`, now default) and a cluster-based one (`clusters=TRUE` or providing your own clusters - the approach from previous versions).
In practice, we observed that both approaches perform well (and better than alternatives).
We suggest using the cluster-based approach when the datasets are segregated into clear clusters, and the random one for the rest (e.g. developmental trajectories).
### Expected proportion of doublets
The expected proportion of doublets has little impact on the score, but a very strong impact on where the threshold will be placed (the thresholding procedure simultaneously minimizes classification error and departure from the expected doublet rate). It is specified through the `dbr` parameter and the `dbr.sd` parameter (the latter specifies the standard deviation of `dbr`, i.e. the uncertainty in the expected doublet rate). For 10x data, the more cells you capture the higher the chance of creating a doublet, and Chromium documentation indicates a doublet rate of roughly 1\% per 1000 cells captures (so with 5000 cells, (0.01\*5)\*5000 = 250 doublets), and the default expected doublet rate will be set to this value (with a default standard deviation of 0.015). Note however that different protocols may create considerably more doublets, and that this should be updated accordingly. If you are unsure about the doublet rate, set `dbr.sd=1` and the thresholding will be entirely based on the misclassification rates.
## Single-cell ATACseq
The `scDblFinder` method can be to single-cell ATACseq (on peak-level counts), however when doing so we recommend using the `aggregateFeatures=TRUE` parameter (see vignette).
In addition, the package includes a reimplementation of the Amulet method from Thibodeau et al. (2021). For more information, see the [ATAC-related vignette](https://plger.github.io/scDblFinder/articles/scATAC.html).
## Comparison with other tools
`scDblFinder` was independently evaluated by Nan Miles Xi and Jingyi Jessica Li in the [addendum](https://arxiv.org/abs/2101.08860) to their excellent [benchmark](https://doi.org/10.1016/j.cels.2020.11.008), where they write that _"scDblFinder achieves the highest mean AUPRC and AUROC values, and it is also the top method in terms of the precision, recall, and TNR under the 10% identification rate."_
The figure below compares some of the methods implemented in this package (in bold) with alternative methods (including the top alternative, `DoubletFinder`):
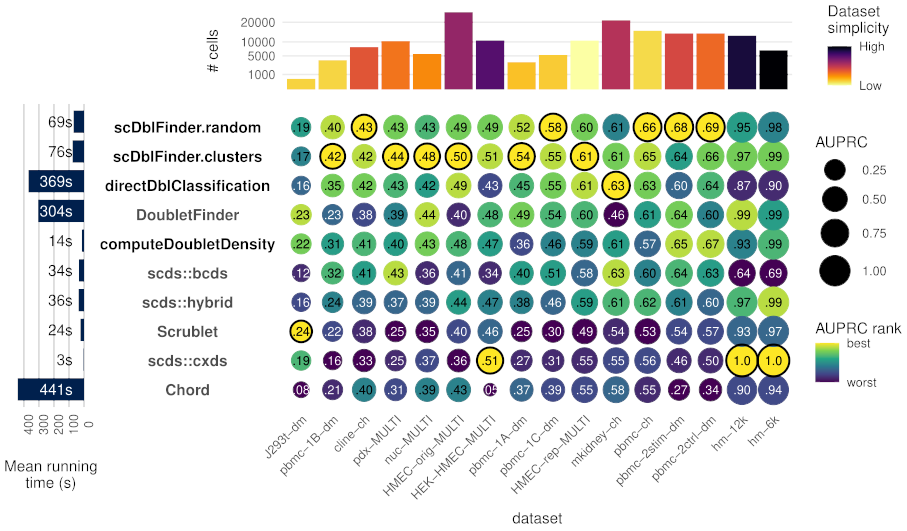
**Figure1:** Accuracy (area under the precision and recall curve) of doublet identification using alternative methods across 16 benchmark datasets from Xi and Li (2020). The colour of the dots indicates the relative ranking for the dataset, while the size and numbers indicate the actual area under the (PR) curve. For each dataset, the top method is circled in black. Methods with names in black are provided in the `scDblFinder` package. Running times are indicated on the left. On top the number of cells in each dataset is shown, and colored by the proportion of variance explained by the first two components (relative to that explained by the first 100), as a rough guide to dataset simplicity.
Rather a python person? You can have a look at [vaeda](https://github.com/kostkalab/vaeda), another doublet finding method which appears to have performances close to those of scDblFinder. Alternatively, run scDblFinder [from the command line](https://plger.github.io/scDblFinder/articles/scDblFinder.html#how-can-i-call-scdblfinder-from-the-command-line).