https://github.com/pwwang/hitype
Hierarchical and high-resolution cell-type identification for single-cell RNA-seq data based on ScType.
https://github.com/pwwang/hitype
cell-type-annotation cell-type-identification scrna-seq-analysis single-cell-rna-seq
Last synced: 2 months ago
JSON representation
Hierarchical and high-resolution cell-type identification for single-cell RNA-seq data based on ScType.
- Host: GitHub
- URL: https://github.com/pwwang/hitype
- Owner: pwwang
- Created: 2023-08-16T05:45:02.000Z (almost 3 years ago)
- Default Branch: master
- Last Pushed: 2023-10-11T22:37:59.000Z (almost 3 years ago)
- Last Synced: 2025-02-04T19:13:10.620Z (over 1 year ago)
- Topics: cell-type-annotation, cell-type-identification, scrna-seq-analysis, single-cell-rna-seq
- Language: R
- Homepage: https://pwwang.github.io/hitype/
- Size: 4.87 MB
- Stars: 0
- Watchers: 2
- Forks: 0
- Open Issues: 0
-
Metadata Files:
- Readme: README.Rmd
- Changelog: NEWS.md
Awesome Lists containing this project
README
---
output: github_document
---
```{r, include = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
fig.path = "man/figures/README-",
out.width = "100%"
)
```
# hitype 
**Hi**erarchical and **hi**gh-resolution cell-type identification for single-cell RNA-seq data inspired by [ScType][1].
## Features
- [x] Compatibility with [ScType][1]
- [x] Hierarchical and high-resolution cell-type identification
- [x] Train weights for your markers with a reference dataset
- [x] Speed optimization
- [x] Support as an R package with unit tests
[1]: https://github.com/IanevskiAleksandr/sc-type
## Installation
You can install the development version of `hitype` like so:
``` r
if (!requireNamespace("devtools", quietly = TRUE)) {
install.packages("devtools")
}
devtools::install_github("pwwang/hitype")
```
## Quick start
### Prepare the dataset
See also
Click to expand
```{r, warning=FALSE, message=FALSE}
pbmc <- pbmc3k.SeuratData::pbmc3k
pbmc[["percent.mt"]] <- Seurat::PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc <- Seurat::NormalizeData(pbmc)
pbmc <- Seurat::FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
pbmc <- Seurat::ScaleData(pbmc, features = rownames(pbmc))
pbmc <- Seurat::RunPCA(pbmc, features = Seurat::VariableFeatures(object = pbmc))
pbmc <- Seurat::FindNeighbors(pbmc, dims = 1:10)
pbmc <- Seurat::FindClusters(pbmc, resolution = 0.5)
pbmc <- Seurat::RunUMAP(pbmc, dims = 1:10)
```
### Use as a Seurat extension
```{r, warning=FALSE, out.width="75%"}
library(hitype)
# Load gene sets
gs <- gs_prepare(hitypedb_pbmc3k)
# Assign cell types
obj <- RunHitype(pbmc, gs)
Seurat::DimPlot(obj, group.by = "hitype", label = TRUE, label.box = TRUE) +
Seurat::NoLegend()
```
Compared to the manual marked cell types:
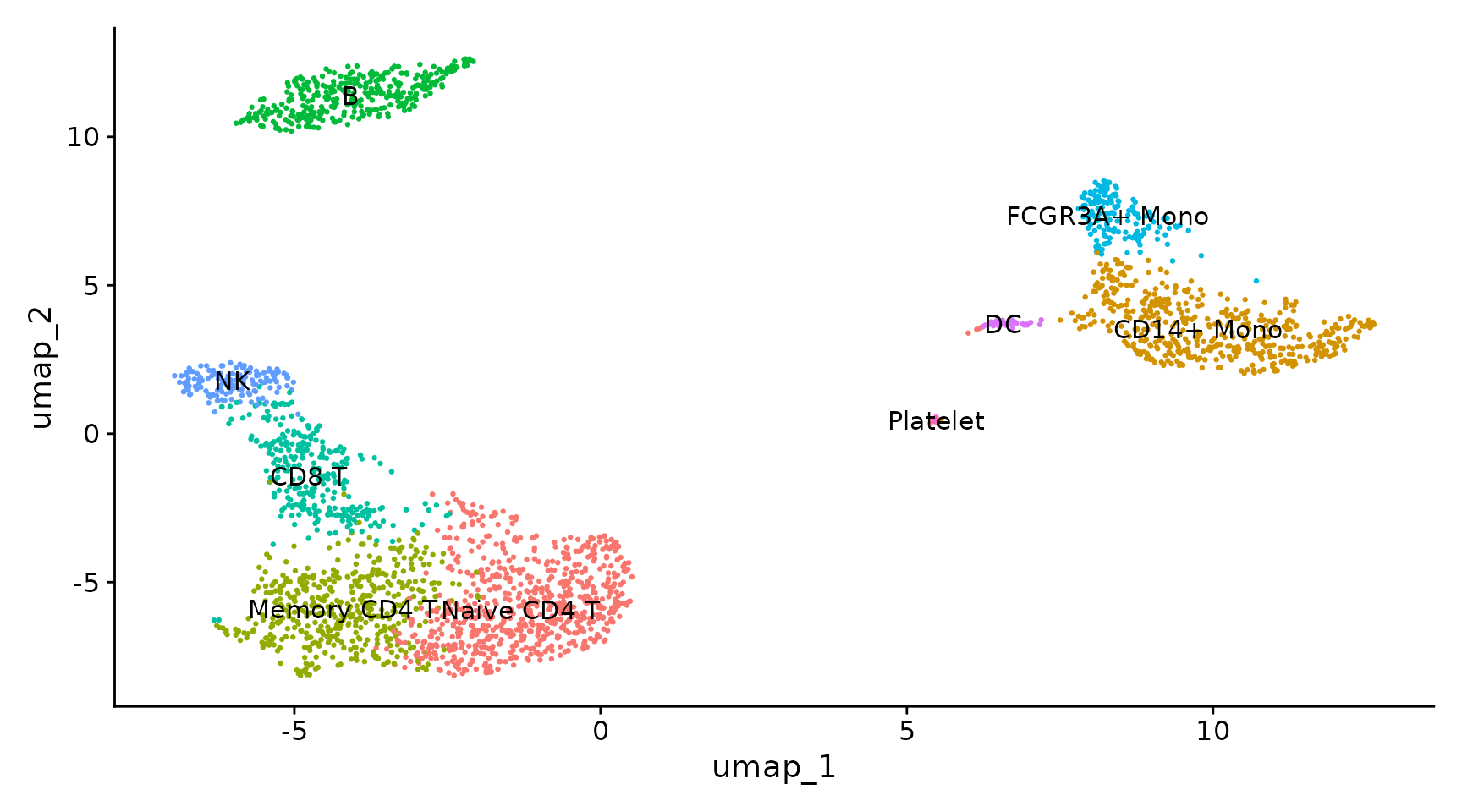{width=75%}
See also
### Use as standalone functions
```{r, warning=FALSE}
scores <- hitype_score(pbmc@assays$RNA@scale.data, gs, scaled = TRUE)
cell_types <- hitype_assign(pbmc$seurat_clusters, scores, gs)
summary(cell_types)
```
You may see that we have exactly the same assignment in the Seurat tutorial:
|Cluster ID | Markers | Cell Type |
|:----------|:--------|:----------|
|0| IL7R, CCR7 | Naive CD4+ T |
|1| CD14, LYZ | CD14+ Mono|
|2| IL7R, S100A4 | Memory CD4+|
|3| MS4A1 | B|
|4| CD8A | CD8+ T|
|5| FCGR3A, MS4A7 | FCGR3A+ Mono|
|6| GNLY, NKG7 | NK|
|7| FCER1A, CST3 | DC|
|8| PPBP | Platelet|
## Documentation