https://github.com/rcedgar/reseek
Protein structure alignment and search algorithm
https://github.com/rcedgar/reseek
bioinformatics bioinformatics-algorithms bioinformatics-tool computational-biology protein-sequences protein-structure search-algorithm
Last synced: over 1 year ago
JSON representation
Protein structure alignment and search algorithm
- Host: GitHub
- URL: https://github.com/rcedgar/reseek
- Owner: rcedgar
- License: gpl-3.0
- Created: 2024-05-21T22:14:10.000Z (about 2 years ago)
- Default Branch: master
- Last Pushed: 2025-03-22T22:11:44.000Z (over 1 year ago)
- Last Synced: 2025-03-22T23:19:57.867Z (over 1 year ago)
- Topics: bioinformatics, bioinformatics-algorithms, bioinformatics-tool, computational-biology, protein-sequences, protein-structure, search-algorithm
- Language: C++
- Homepage: https://drive5.com/reseek
- Size: 24 MB
- Stars: 59
- Watchers: 2
- Forks: 3
- Open Issues: 4
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
README

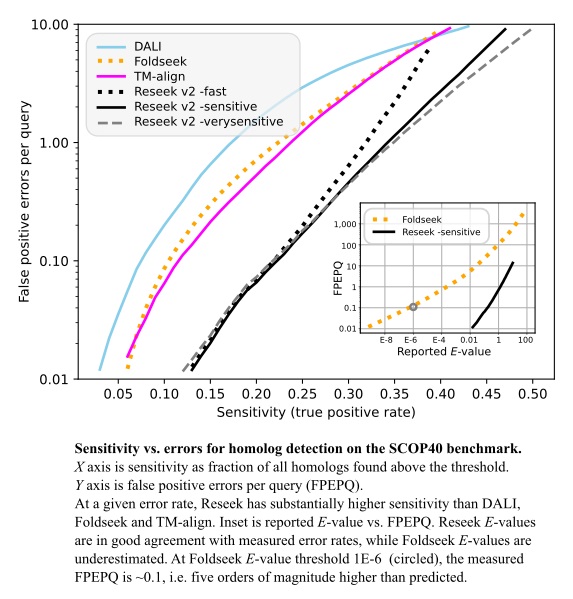
[Reseek](http://drive5.com/reseek) is a protein structure search and alignment algorithm which improves sensitivity in protein homolog detection
compared to state-of-the-art methods including DALI, TM-align and Foldseek with similar speed to Foldseek.
Reseek is based on sequence alignment where each residue in the protein backbone is represented by a
letter in a novel “mega-alphabet” of 85,899,345,920 (∼1011) distinct states.
Method sensitivity was measured on the SCOP40 benchmark using superfamily as the truth standard, focusing
on the regime with false-positive error rates <10 per query, corresponding to E<10 for an ideal E-value.
[ ](https://www.youtube.com/watch?v=BzIgqdm9xDs)
](https://www.youtube.com/watch?v=BzIgqdm9xDs)
### Command line
-search # Alignment (e.g. DB search, pairwise, all-vs-all)
-convert # Convert file formats (e.g. create DB)
-alignpair # Pair-wise alignment and superposition
Search against database
reseek -search STRUCTS -db STRUCTS -output hits.txt
# STRUCTS specifies structure(s), see below
Recommended format for large database is .bca, e.g.
reseek -convert /data/PDB_mirror/ -bca PDB.bca
Align and superpose two structures
reseek -alignpair 1XYZ.pdb -input2 2ABC.pdb
-aln FILE # Sequence alignment (text)
-output FILE # Rotated 1XYZ (PDB format)
All-vs-all alignment
reseek -search STRUCTS -output hits.txt
Output options for -search
-aln FILE # Alignments in human-readable format
-output FILE # Hits in tabbed text format
-columns name1+name2+name3...
# Output columns, names are
# query Query label
# target Target label
# qlo Start of aligment in query
# qhi End of aligment in query
# tlo Start of aligment in target
# thi End of aligment in target
# ql Query length
# tl Target length
# pctid Percent identity of alignment
# cigar CIGAR string
# evalue You can guess this one
# aq AQ (aln. qual., 0 to 1, >0.5 suggests homology)
# qrow Aligned query sequence with gaps (local)
# trow Aligned target sequence with gaps (local)
# qrowg Aligned query sequence with gaps (global)
# trowg Aligned target sequence with gaps (global)
# std query+target+qlo+qhi+ql+tlo+thi+tl+pctid+evalue
# default aq+query+target+evalue
Search and alignment options
-fast, -sensitive or -verysensitive # Required
-evalue E # Max E-value (default 10 unless -verysensitive)
-omega X # Omega accelerator (floating-point)
-minu U # K-mer accelerator (integer)
-gapopen X # Gap-open penalty (floating-point >= 0)
-gapext X # Gap-extend penalty (floating-point >= 0)
-dbsize D # DB size (nr. chains) for E-value (default actual size)
Convert between file formats
reseek -convert STRUCTS [one or more output options]
-cal FILENAME # .cal format, text with a.a. and C-alpha x,y,z
-bca FILENAME # .bca format, binary .cal, recommended for DBs
-fasta FILENAME # FASTA format
Create input for Muscle-3D multiple structure alignment:
reseek -pdb2mega STRUCTS -output structs.mega
STRUCTS argument is one of:
NAME.cif or NAME.mmcif # PDBx/mmCIF file
NAME.pdb # Legacy format PDB file
NAME.cal # C-alpha tabbed text format with chain(s)
NAME.bca # Binary C-alpha, recommended for larger DBs
NAME.files # Text file with one STRUCT per line,
# may be filename, directory or .files
DIRECTORYNAME # Directory (and its sub-directories) is searched
# for known file types including .pdb, .files etc.
Other options:
-log FILENAME # Log file with errors, warnings, time and memory.
-threads N # Number of threads, default number of CPU cores.
#### Build from source on Linux x86
cd src/; chmod +x build_linux_x86.bash ; ./build_linux_x86.bash
#### Build from source on OSX x86
cd src/ ; chmod +x build_osx_x86.bash ; ./build_osx_x86.bash
#### Build from source on Windows
Load `reseek.vcxproj` into Microsoft Visual Studio and use the Build command.
#### Static link warning
Don't worry about a warning something like this, it's expected:
warning: Using 'dlopen' in statically linked applications requires
at runtime the shared libraries from the glibc version used for linking
### More documentation
[https://drive5.com/reseek](https://drive5.com/reseek)
### Reference
Edgar, Robert C. (2024) "Sequence alignment using large protein structure alphabets improves sensitivity to remote homologs" [https://www.biorxiv.org/content/10.1101/2024.05.24.595840v2](https://www.biorxiv.org/content/10.1101/2024.05.24.595840v2)
### SCOP40 benchmark code and results
https://github.com/rcedgar/reseek_bench