https://github.com/reskejak/ATAC-seq
Basic workflow for ATAC-seq analysis
https://github.com/reskejak/ATAC-seq
atac-seq r shell unix
Last synced: about 1 year ago
JSON representation
Basic workflow for ATAC-seq analysis
- Host: GitHub
- URL: https://github.com/reskejak/ATAC-seq
- Owner: reskejak
- Created: 2018-03-30T13:50:43.000Z (over 8 years ago)
- Default Branch: master
- Last Pushed: 2022-01-14T13:45:27.000Z (over 4 years ago)
- Last Synced: 2024-02-14T03:31:30.240Z (over 2 years ago)
- Topics: atac-seq, r, shell, unix
- Language: R
- Homepage:
- Size: 123 KB
- Stars: 54
- Watchers: 4
- Forks: 27
- Open Issues: 3
-
Metadata Files:
- Readme: README.md
Awesome Lists containing this project
- Awesome-Bioinformatics-Benchmarks - Github
README
# ATAC-seq
These scripts correspond to a (differential) ATAC-seq analysis workflow as described in [our recent report](https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-020-00342-y). It is largely based on the pipeline developed by [Anshul Kundaje's group (Stanford) and the ENCODE project](https://www.encodeproject.org/pipelines/ENCPL792NWO/).
If you use this methodology, please cite the following paper along with corresponding pipeline dependencies:
Jake J. Reske, Mike R. Wilson, and Ronald L. Chandler. 2020. ATAC-seq normalization method can significantly affect differential accessibility analysis and interpretation. *Epigenetics & Chromatin* **13**: 22.
Attempt to run each command individually or in blocks after editing to match your own data architecture.
### ATACseq_workflow.txt
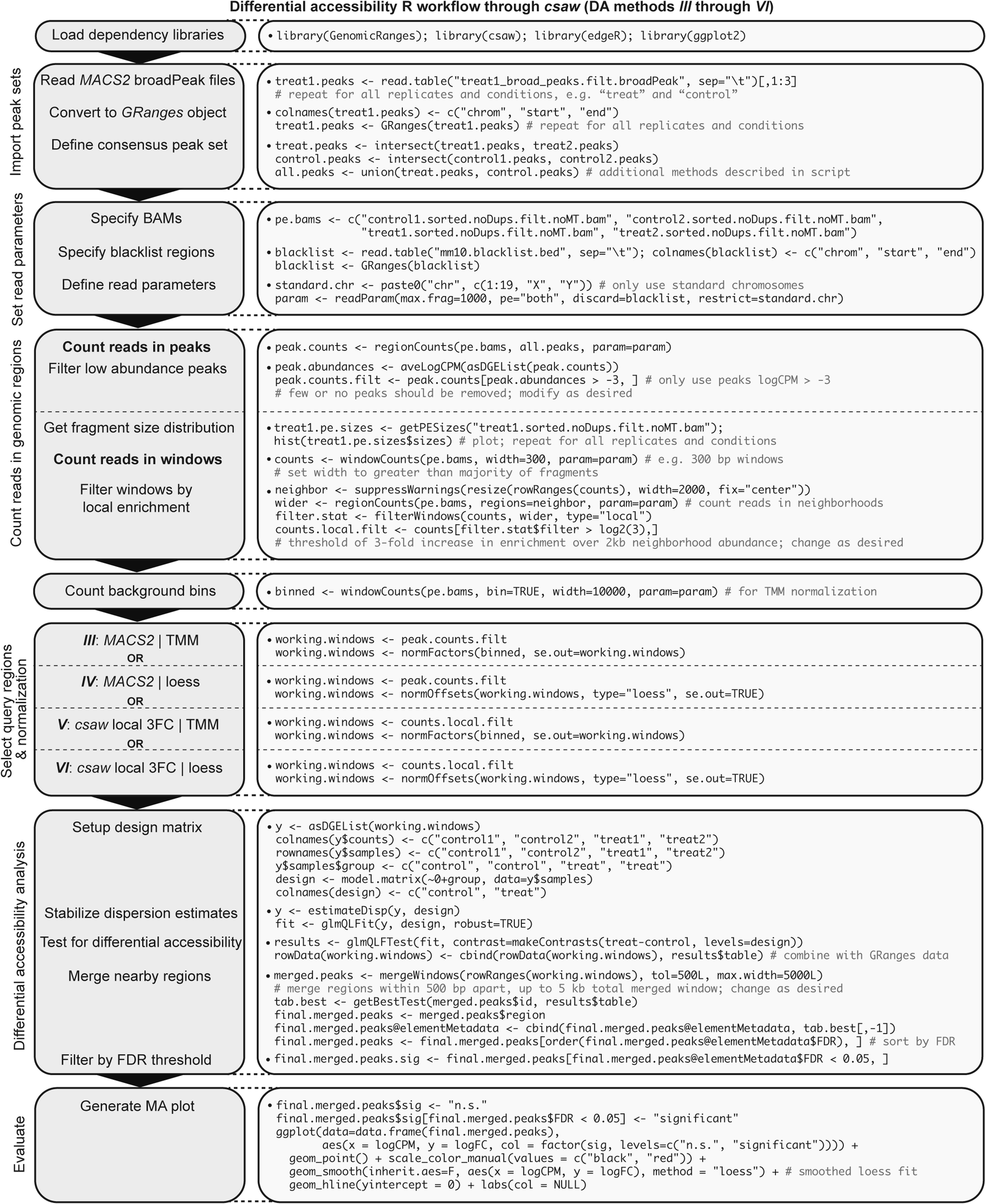
**Generalized ATAC-seq data processing workflow intended for comparative analysis.** Stepwise bioinformatics process and example commands for analyzing ATAC-seq data from raw reads to calling peaks for downstream differential accessibility analysis. Consider “treat1” as an example mouse ATAC-seq Illumina paired-end library. Blue text denotes optional or conditional steps dependent on experimental design and desired output. Users seeking only to discover replicate-concordant accessible regions in a singular cell state may wish to call naïve overlapping peaks, though this step is not necessary for differential accessibility analysis.
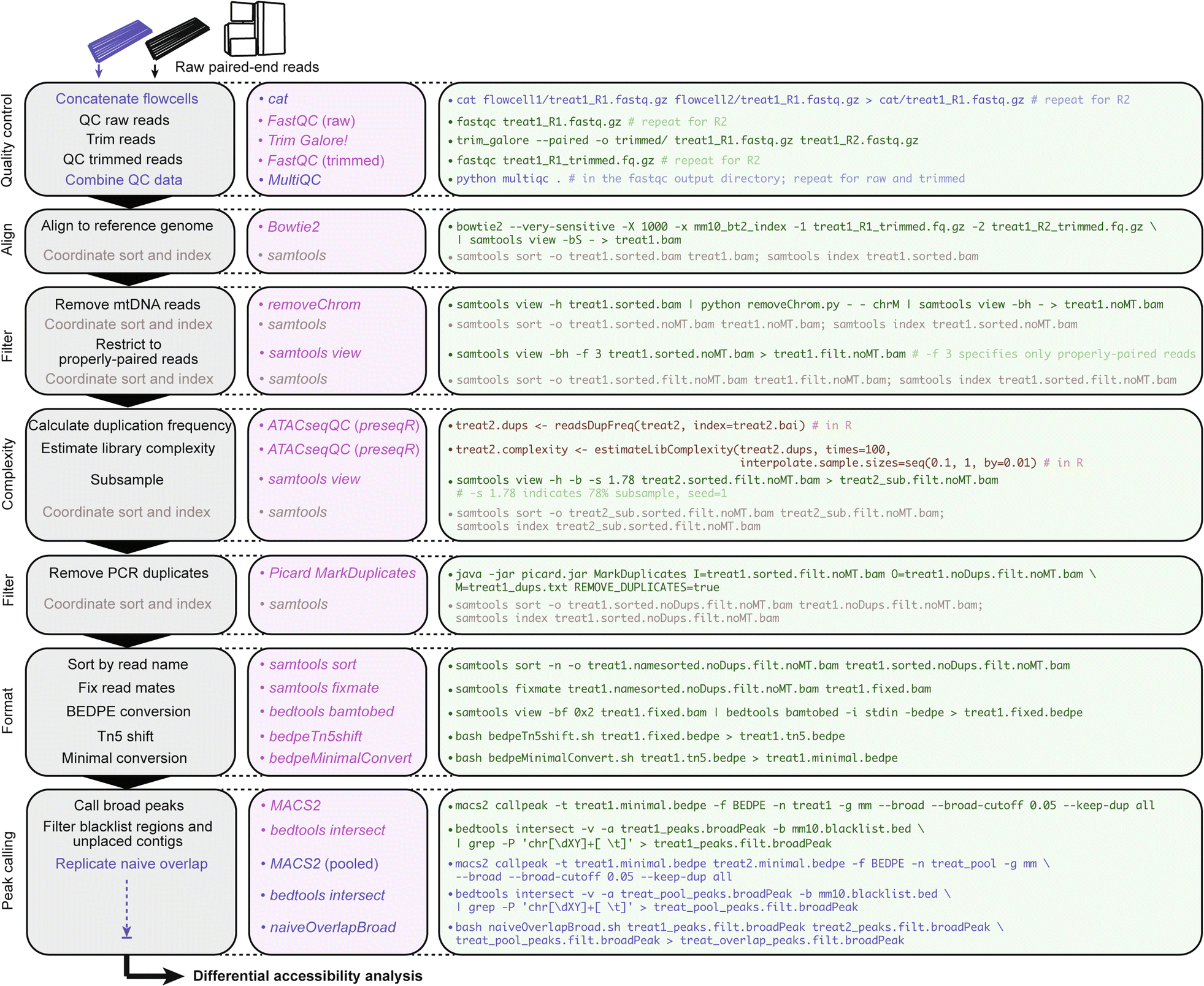
### csaw_workflow.R
***csaw* workflow for multiple differential accessibility analyses in R.** Consider an experimental design with *n* = 2 biological replicates from two conditions: “treat” and “control”. Describes implementation of two possible normalization methods and use of either *MACS2* peaks or *de novo* locally enriched windows as query regions for output comparison; see [*csaw* manual](https://bioconductor.org/packages/release/bioc/html/csaw.html) for additional normalization frameworks. ***Note: updates to the behavior of certain csaw functions have required slight compatibility changes to the commands described graphically, so please reference the latest R script.***