https://github.com/slowikj/seqr
fast and comprehensive k-mer counting package
https://github.com/slowikj/seqr
bioinformatics bioinformatics-tool dna-processing feature-engineering feature-extraction genomics hashing hashing-algorithms k-mer k-mer-counting kmer kmer-counting kmer-frequency-count kmers ngram ngrams protein-sequences rcpp rcppparallel rpackage
Last synced: about 1 year ago
JSON representation
fast and comprehensive k-mer counting package
- Host: GitHub
- URL: https://github.com/slowikj/seqr
- Owner: slowikj
- Created: 2019-11-14T06:20:51.000Z (over 6 years ago)
- Default Branch: master
- Last Pushed: 2021-09-27T16:40:05.000Z (almost 5 years ago)
- Last Synced: 2025-04-03T08:03:37.322Z (over 1 year ago)
- Topics: bioinformatics, bioinformatics-tool, dna-processing, feature-engineering, feature-extraction, genomics, hashing, hashing-algorithms, k-mer, k-mer-counting, kmer, kmer-counting, kmer-frequency-count, kmers, ngram, ngrams, protein-sequences, rcpp, rcppparallel, rpackage
- Language: C++
- Homepage: https://slowikj.github.io/seqR/
- Size: 1.72 MB
- Stars: 18
- Watchers: 2
- Forks: 1
- Open Issues: 7
-
Metadata Files:
- Readme: README.Rmd
Awesome Lists containing this project
README
---
output: github_document
---
```{r setup, include=FALSE}
knitr::opts_chunk$set(
echo = TRUE,
collapse = TRUE,
comment = "#>",
out.width = "100%"
)
```
```{r, include = FALSE}
library(seqR)
```
# seqR - fast and comprehensive k-mer counting package
[](https://cran.r-project.org/package=seqR)
[](https://github.com/slowikj/seqR/actions)
[](https://lifecycle.r-lib.org/articles/stages.html#stable)
[](https://www.gnu.org/licenses/gpl-3.0)
[](https://codecov.io/github/slowikj/seqR?branch=master)
[](https://www.code-inspector.com/project/23909/status/svg)
[](https://www.code-inspector.com/project/23909/score/svg)
## About
`seqR` is an R package for fast k-mer counting. It provides
* **highly optimized** (the core algorithm is written in C++)
* **in-memory**
* **probabilistic** (with configurable dimensionality of a hash value
used for storing k-mers internally),
* **multi-threaded** (with a configurable size of the batch of sequences (`batch_size`) to process in a single step. If `batch_size` equals 1, the multi-threaded mode is disabled, which potentially causes a longer computation time)
implementation that supports
* **various variants of k-mers** (contiguous, gapped, and positional counterparts)
* **all biological sequences** (e.g., nucleic acids and proteins)
Moreover, the result optimizes memory consumption by the application of **sparse matrices**
(see [package Matrix](https://CRAN.R-project.org/package=Matrix)),
compatible with machine learning packages
such as [ranger](https://CRAN.R-project.org/package=ranger)
and [xgboost](https://CRAN.R-project.org/package=xgboost).
## How to...
### How to install
To install `seqR` from CRAN:
```{r, eval=FALSE}
install.packages("seqR")
```
Alternatively, if you want to use the latest development version:
```{r, eval=FALSE}
# install.packages("devtools")
devtools::install_github("slowikj/seqR")
```
### How to use
The package provides two functions that facilitate k-mer counting
* `count_kmers` (used for counting k-mers of one type)
* `count_multimers` (a wrapper of `count_kmers`, used for counting k-mers of many types in a single invocation of the function)
and one function used for custom processing of k-mer matrices:
* `rbind_columnwise` (a helper function used for merging several k-mer matrices that do not have same sets of columns)
To learn more, see [features overview vignette](https://slowikj.github.io/seqR/articles/features-overview.html)
and [reference](https://slowikj.github.io/seqR/reference/index.html).
#### Examples
##### counting 5-mers
```{r}
count_kmers(sequences=c("AAAAAVVAVFF", "DFGSADFGSA"),
k=5)
```
##### counting gapped 5-mers with gaps (0, 1, 0, 2) (XX_XX__X)
```{r}
count_kmers(sequences=c("AAAAAVVAVFF", "DFGSADFGSA"),
kmer_gaps=c(0, 1, 0, 2))
```
##### counting 1-mers and 2-mers
```{r}
data(CsgA)
CsgA[1L:2]
count_multimers(sequences=CsgA,
k_vector = c(1, 2))
```
### How to cite
For citation type:
```{r, eval=FALSE}
citation("seqR")
```
or use:
Jadwiga Słowik and Michał Burdukiewicz (2021). seqR: fast and comprehensive k-mer counting package. R package version 1.0.0.
## Benchmarks
The `seqR` package has been compared with other existing k-mer counting R packages:
[biogram](https://CRAN.R-project.org/package=biogram),
[kmer](https://CRAN.R-project.org/package=kmer),
[seqinr](https://CRAN.R-project.org/package=seqinr),
and [biostrings](https://bioconductor.org/packages/Biostrings).
All benchmark experiments have been performed using Intel Core i7-6700HQ 2.60GHz 8 cores, using the [microbenchmark](https://CRAN.R-project.org/package=microbenchmark) R package.
### Contiguous k-mers
#### Changing k
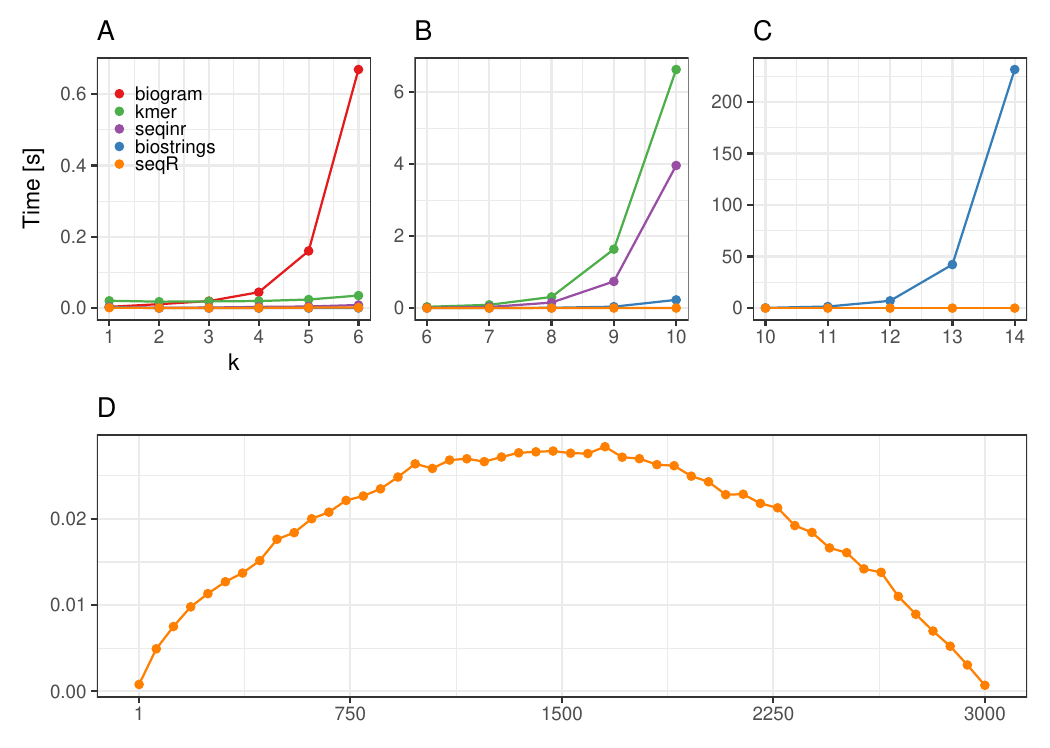
The input consists of one `DNA` sequence of length `3 000`.
#### Changing the number of sequences
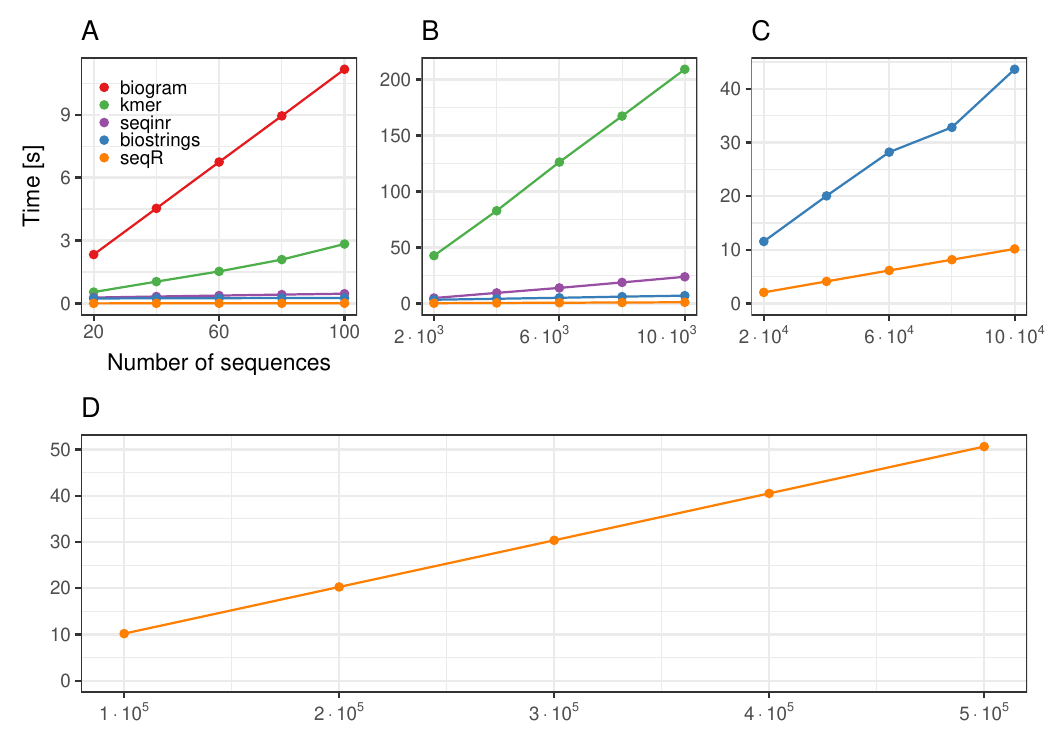
Each `DNA` sequence has `3 000` elements, `contiguous 5-mer` counting.
### Gapped k-mers
#### Changing the first contiguous part of a k-mer
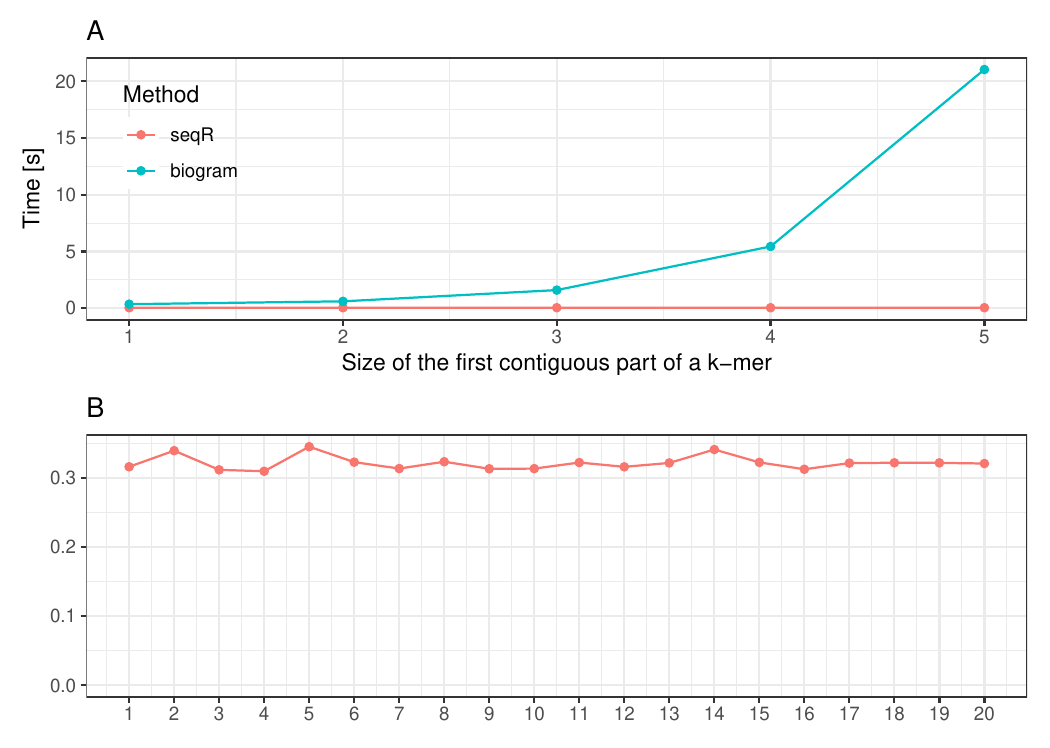
The input consists of one `DNA` sequence of length `1 000 000`. `Gapped 5-mers` counting with base gaps `(1, 0, 0, 1)`.
#### Changing the first gap size
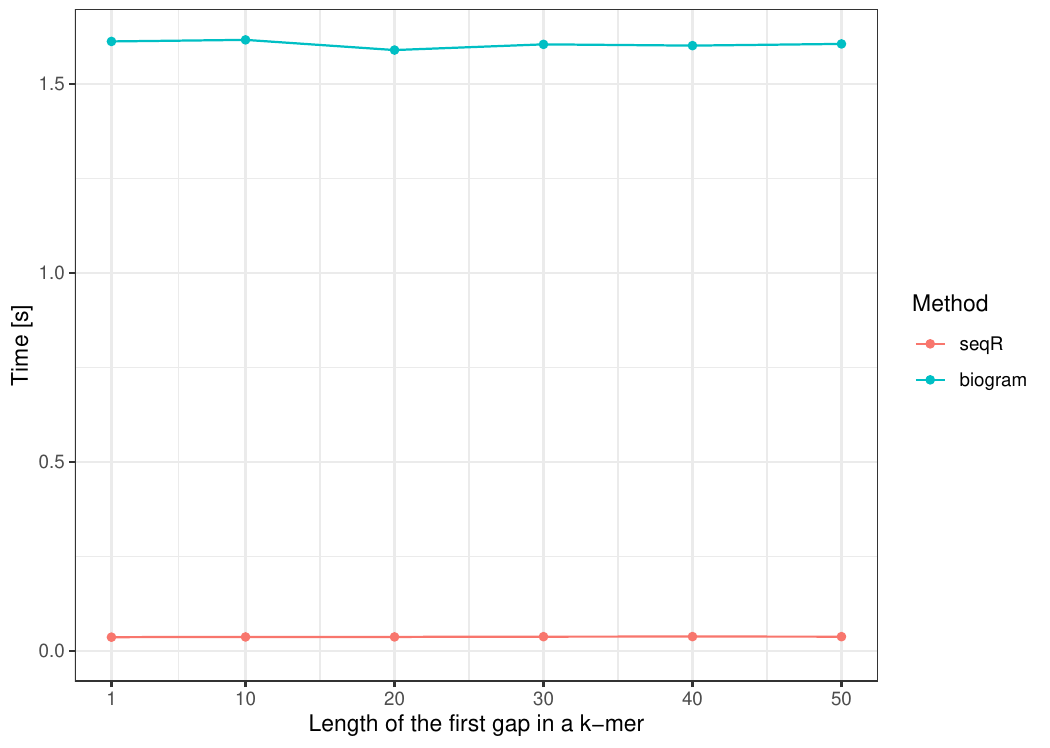
The input consists of one `DNA` sequence of length `100 000`. `Gapped 5-mers` counting with base gaps `(1, 0, 0, 1)`.