https://github.com/stracquadaniolab/quick-rnaseq-nf
A basic and quick workflow for differential expression analysis
https://github.com/stracquadaniolab/quick-rnaseq-nf
nextflow rnaseq
Last synced: 4 months ago
JSON representation
A basic and quick workflow for differential expression analysis
- Host: GitHub
- URL: https://github.com/stracquadaniolab/quick-rnaseq-nf
- Owner: stracquadaniolab
- License: mit
- Created: 2021-09-29T14:42:25.000Z (about 4 years ago)
- Default Branch: master
- Last Pushed: 2022-06-17T12:25:37.000Z (over 3 years ago)
- Last Synced: 2025-03-01T16:47:54.623Z (7 months ago)
- Topics: nextflow, rnaseq
- Language: Nextflow
- Size: 5.92 MB
- Stars: 1
- Watchers: 1
- Forks: 1
- Open Issues: 0
-
Metadata Files:
- Readme: readme.md
- License: LICENSE
Awesome Lists containing this project
README
# quick-rnaseq-nf


[](https://doi.org/10.5281/zenodo.6656442)
A basic and quick workflow for differential expression analysis.
## Overview
[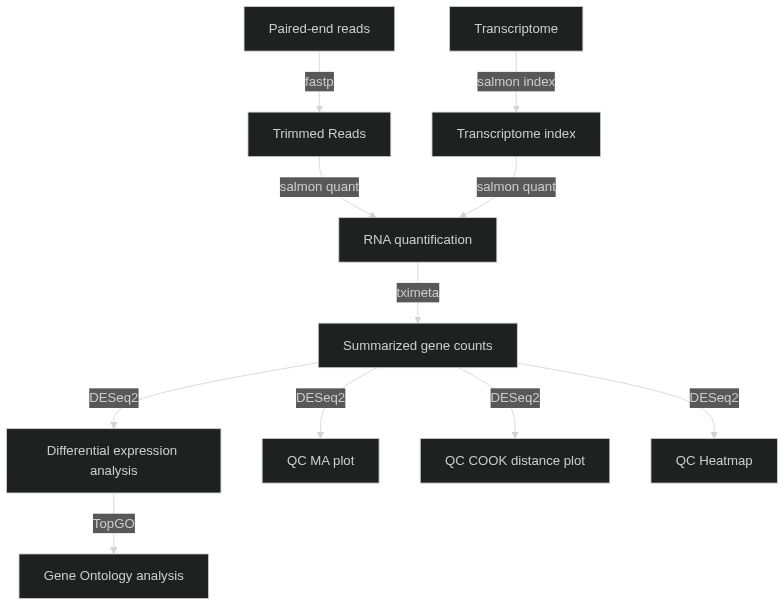](https://mermaid.live/edit#pako:eNp9ks1OwzAQhF9l5TM90GORkNqkf0JQIL0lPaziTWsR28HeiBbSd8dtXAlQhQ-W9c14vBr5S5RWkhiJqrYf5Q4dwzq9KwyENc7XTmlNEl4Jpd_0dBIoGl861bDVBMpI2kctyV-fxvDeomFVqRJZWROlNM9ardGpz5C3JUNQ2tbwJXWap6qqyFG4iTXQvnHkfbgOaLA-eHUxzvKXBB7H0NSWI5qfULJaPYBUntGU9FNdnNQFIWtsIlrm89MAK8O2ttvD3yf6PbvNn1E5kgMyEty5gcHgHroKPTcdjCHWlA1_N7KBs81jrcP453o6mMRKf2nnojpIYtDkPzHpRd4rTYxdGlvtaTrN6H3YwewanV-Di1h7D9e2ma-65dXMqbgRmpxGJcM3-Tp5CsE70lSIUThKdG-FKMwx-NpGItNUKrZOjCqsPd0IbNlmB1OKEbuWLqZU4dahjq7jN65wzq0)
## Configuration
- `codename`: mnemonic codename for the run (default: 'quick-rnaseq')
- `outdir`: directory where to store the results (default: './results')
- `experiment.samplesheet`: CSV file describing samples and conditions (required).
- `experiment.contrasts`: contrasts for differential expression analysis in the
format [['case1', 'control'],['case2','control']], which performs the analysis
of case1 vs control samples, and case2 vs control samples. (required)
- `transcriptome.reference`: transcriptome reference file. Gencode recommended (required. GZ format)
- `transcriptome.decoys`: reference genome file. Gencode recommended (required. GZ format)
- `fastp.args`: options for reads trimming using fastp. (default: '')
- `salmon.index.args`: options for Salmon index, e.g. '--gencode' for Gencode transcritomes.
- `salmon.quant.libtype`: library type for quantification (default: 'A', Salmon infers lib type)
- `salmon.quant.args`: Salmon options. (default: '--validateMappings --gcBias')
- `summarize_to_gene.counts_from_abundance`: infer counts from abundances using tximeta (default: 'no')
- `summarize_to_gene.organism_db`: Bioconductor organism package for annotation.
Currently supporting `org.Hs.eg.db` for Human and `org.Mm.eg.db` for mouse and
(default: 'org.Hs.eg.db')
- `qc.pca.transform`: counts transformation for PCA analysis (see DESeq2. default: 'rlog')
- `qc.ma.lfc_threshold`: log fold-change threshold for MA plot (see DESeq2. default: 0)
- `qc.sample.transform`: counts transformation for PCA analysis (see DESeq2. default: 'rlog')
- `dge.lfc_threshold`: log fold-change threshold for differential expression analysis (see DESeq2. default: 0)
- `dge.fdr`: false discovery rate threshold to be used with implicit filtering (see DESeq2. default: 0.05)
- `gene_ontology.organism_db`: Bioconductor organism package for annotation.
Currently supporting `org.Hs.eg.db` for Human and `org.Mm.eg.db` for mouse and
(default: 'org.Hs.eg.db')
- `gene_ontology.gene_id`: type of gene id used for the analysis (default: 'ensembl')
- `gene_ontology.remove_gencode_version`: remove Gencode version from gene id (default: 'yes')
- `gene_ontology.fdr`: false discovery rate threshold (default: 0.05)
## Running the workflow
### Install or update the workflow
```bash
nextflow pull stracquadaniolab/quick-rnaseq-nf
```
### Run the analysis with test data and Docker
```bash
nextflow run stracquadaniolab/quick-rnaseq-nf -profile test,docker
```
### Run the analysis with test data and Singularity
```bash
nextflow run stracquadaniolab/quick-rnaseq-nf -profile test,singularity
```
### Run the analysis with test data, Singularity and Slurm
```bash
nextflow run stracquadaniolab/quick-rnaseq-nf -profile test,singularity,slurm
```
### Run the analysis on human data with Singularity and Slurm
Prepare a `samplesheet.csv` file as follows:
```
sample,read1,read2,condition
6C_REP1,data/RF01_6C1_R1_001.fastq.gz,data/RF01_6C1_R2_001.fastq.gz,control
6C_REP2,data/RF01_6C2_R1_001.fastq.gz,data/RF01_6C2_R2_001.fastq.gz,case
```
Please note that the header is required and it is case sensitive.
Prepare a `nextflow.config` file as follows:
```
params {
// experiment information
experiment.samplesheet = "./samplesheet.csv"
experiment.contrasts = [['case1', 'control'],['case2','control']]
// transcriptome information
transcriptome.reference = "gencode.v40.transcripts.fa.gz"
transcriptome.decoys = "GRCh38.primary_assembly.genome.fa.gz"
}
```
Now you can run `quick-rnaseq` as follows:
```bash
nextflow run stracquadaniolab/quick-rnaseq-nf -profile singularity,slurm
```
## Results
- `results/analysis/dge-.csv`: file with the differential expression analysis results for a given contrast.
- `results/analysis/go-.csv`: file with the GO analysis results for a given contrast.
- `results/dataset/summarized-experiment.rds`: DESeqDataset object with all experimental information (e.g. gene counts)
- `results/qc`: quality control report
- `results/quantification/`: Salmon quantification folders for each sample.
## Authors
- Giovanni Stracquadanio, giovanni.stracquadanio@ed.ac.uk