https://github.com/yufree/mzrtsim
Raw data and peaks list simulation for GC/LC-MS based data
https://github.com/yufree/mzrtsim
metabolomics mzml simulation
Last synced: 4 months ago
JSON representation
Raw data and peaks list simulation for GC/LC-MS based data
- Host: GitHub
- URL: https://github.com/yufree/mzrtsim
- Owner: yufree
- Created: 2018-04-12T22:56:39.000Z (about 8 years ago)
- Default Branch: master
- Last Pushed: 2025-08-17T22:31:58.000Z (10 months ago)
- Last Synced: 2025-08-25T05:01:02.805Z (10 months ago)
- Topics: metabolomics, mzml, simulation
- Language: R
- Homepage: https://yufree.github.io/mzrtsim/
- Size: 58.1 MB
- Stars: 3
- Watchers: 2
- Forks: 1
- Open Issues: 0
-
Metadata Files:
- Readme: README.Rmd
Awesome Lists containing this project
README
---
output: github_document
---
```{r, include = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
fig.path = "man/figures/README-",
out.width = "100%"
)
```
# mzrtsim
[](https://github.com/yufree/mzrtsim/actions/workflows/R-CMD-check.yaml)
[](https://lifecycle.r-lib.org/articles/stages.html#stable)
The goal of mzrtsim is to make raw data and features table simulation for LC/GC-MS based data
## Citation
Yu, M.; Philip, V. Mzrtsim: Raw Data Simulation for Reproducible Gas/Liquid Chromatography–Mass Spectrometry-Based Nontargeted Metabolomics Data Analysis. Anal. Chem. 2025, 97 (32), 17309–17314. https://doi.org/10.1021/acs.analchem.5c01213.
## Installation
You can install the development version from [GitHub](https://github.com/) with:
```{r eval=FALSE}
# install.packages("remotes")
remotes::install_github("yufree/mzrtsim")
```
## Raw Data simulation
MS1 full scan data has been proved more complex than theoretical prediction. Recently study showed that soft ionization will also contain [fragment ions](https://www.nature.com/articles/s41596-023-00803-0) for structure identification and contain lots of [redundant peaks](https://pubs.acs.org/doi/10.1021/acs.analchem.7b02380). As shown in the following figure, MS1 full scan data could contain different types of ions from the same compound (red). Some peaks we could figure out the sources while some peaks might be hard to interpret. In this case, mzrtsim package will use experimental data instead of predicted peaks from the compound formula to show the complexity of MS1 spectra.
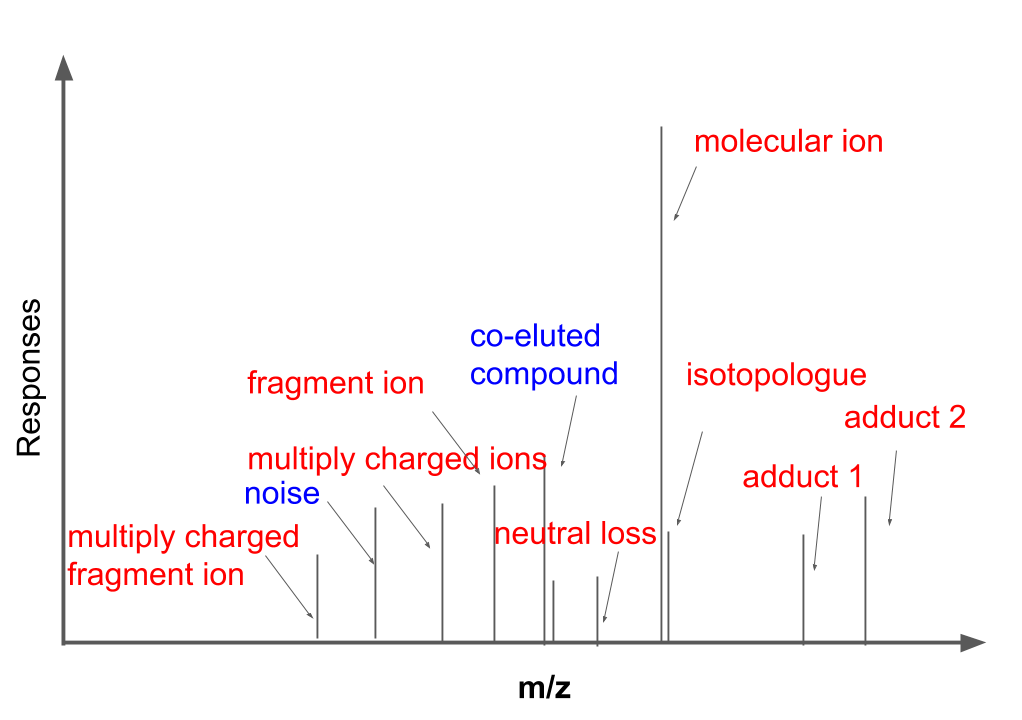
You could use `simmzml` to generate one mzML file.
```{r eval=FALSE}
library(mzrtsim)
data("monams1")
simmzml(db=monams1, name = 'test')
```
You will find `test.mzML` and corresponding `test.csv` with m/z, retention time and compound name of the peaks. Here the `monams1` and `monahrms1` is from the MS1 data of MassBank of North America (MoNA) and could be downloaded from their [website](https://mona.fiehnlab.ucdavis.edu/downloads). You could also use `hmdbcms` to simulate EI source data extracted from [HMDB](https://hmdb.ca/downloads). Here we only use the MS1 full scan data for simulation.
### Data extraction
For HMDB, you need to download their "GC-MS Spectra Files (XML) - Experimental" data [here](https://hmdb.ca/downloads). Then you need the following R code to construct the database for this package.
```{r eval=FALSE}
xmlpath <- list.files('hmdb_experimental_cms_spectra/',full.names = T,recursive = T)
library(xml2)
getcms <- function(i){
tree = xml2::read_xml(xmlpath[i])
accession = xml_text(xml2::xml_find_all(tree,'/c-ms/database-id'))
rti = as.numeric(xml_text(xml2::xml_find_all(tree,'/c-ms/retention-index')))
prec = as.numeric(xml_text(xml2::xml_find_all(tree,'/c-ms/derivative-exact-mass')))
formula = xml_text(xml2::xml_find_all(tree,'/c-ms/derivative-formula'))
ins = xml_text(xml2::xml_find_all(tree,'/c-ms/notes'))
ins2 = xml_text(xml2::xml_find_all(tree,'/c-ms/instrument-type'))
mode = xml_text(xml2::xml_find_all(tree,'/c-ms/ionization-mode'))
idms = xml_text(xml2::xml_find_all(tree,'/c-ms/id'))
instr = xml_text(xml2::xml_find_all(tree,'instrument-type'))
masscharge = as.numeric(xml_text(xml2::xml_find_all(tree,'/c-ms/c-ms-peaks/c-ms-peak/mass-charge')))
intensity = as.numeric(xml_text(xml2::xml_find_all(tree,'/c-ms/c-ms-peaks/c-ms-peak/intensity')))
idx <- intensity!=0
mz <- list(name=accession, idms=idms, ionmode=mode, prec = prec, formula = formula, np = length(masscharge[idx]), rti = rti, instr = ins2, msm = ins, spectra=cbind.data.frame(mz=masscharge[idx],ins=intensity[idx]))
return(mz)
}
library(parallel)
hmdbcms <- mcMap(getcms,1:length(xmlpath), mc.cores = 4)
saveRDS(hmdbcms,'hmdbcms.RDS')
```
For MoNA, you can download the "LC-MS Spectra" data in format of "msp" from this [website](https://mona.fiehnlab.ucdavis.edu/downloads). Then we need to filter the MS1 data to generate database for LC-MS and subset data base for LC-HRMS.
```{r eval=FALSE}
# you need to install enviGCMS package by 'install.packages("enviGCMS")'.
msp <- enviGCMS::getMSP('MoNA-export-LC-MS_Spectra.msp')
idx <- sapply(msp, function(x) grepl('MS1',x$msm))
idx1 <- sapply(idx, function(x) length(x)>0)
monams1 <- msp[idx1]
idx2 <- sapply(monams1, function(x) grepl('MS1',x$msm))
monams1 <- monams1[idx2]
idx3 <- sapply(monams1, function(x) x$instr )
idx4 <- sapply(idx3, function(x) length(x)>0)
monams12 <- monams1[idx4]
saveRDS(monams12,'monams1.RDS')
idx5 <- sapply(monams12, function(x) x$instr )
monams13 <- monams12[grepl('FT|TOF',idx5)]
idx5 <- sapply(monams13, function(x) x$instr )
nn <- sapply(monams13,function(x) x$np)
median(as.numeric(nn))
mean(as.numeric(nn))
saveRDS(monams13,'monahrms1.RDS')
```
If you have your own database in "msp" format, you can generate the custom database by the following code:
```{r eval=FALSE}
msp <- enviGCMS::getMSP('MoNA-export-LC-MS_Spectra.msp')
saveRDS(msp,'custom.RDS')
```
To use the "*.RDS" database, you need to load them and refer their name for 'simmzml' function.
```{r eval=FALSE}
custom <- readRDS('custom.RDS')
simmzml(db=custom, name = 'test')
```
To select certain compound for review/simulation, you could use name or inchikey.
```{r}
data(monahrms1)
# if you know the compound name
compoundname <- 'Triacylglycerol 14:0-16:0-16:0'
databasename <- sapply(monahrms1,function(x) x$name)
compoundidx <- databasename%in%compoundname
compound <- monahrms1[compoundidx]
compound
# if you know the inchikey
compoundinchikey <- c('KQSFNXMDCOFFGW-GNDIVNLPSA-N','WRYJYFCCMSVEPQ-ORAXXRKOSA-N')
databaseinchikey <- sapply(monahrms1,function(x) x$inchikey)
compoundidx <- databaseinchikey%in%compoundinchikey
compound <- monahrms1[compoundidx]
compound
```
### Multiple files with experiment design
You could stimulate two groups of raw data with different peak heights for the same compounds. Retention time will follow a uniform distribution. 100 compounds could be selected randomly and base peaks' signal to noise ratio could be sampled from 100 to 1000. When signal to noise ratio is fixed for all samples, the peaks height will still be the only changed parameters for both groups. Each group contain 10 samples and 30% compounds are changed between case and control groups.
```{r eval=FALSE}
dir.create('case')
dir.create('control')
# set different peak height for 100 compounds
ph1 <- c(rep(5,30),rep(10,40),rep(15,30))
ph2 <- c(rep(5,20),rep(10,30),rep(15,50))
# set retention time for 100 compounds. When the peak width is set as a single value, the simulated base peak width will be from a poisson distribution with this value as lambda.
rt <- seq(10,590,length.out=100)
set.seed(1)
# select compounds from database
compound <- sample(c(1:4000),100)
set.seed(2)
# select signal to noise ratio for a larger dynamic range
sn <- sample(c(100:10000),100)
for(i in c(1:10)){
simmzml(name=paste0('case/case',i),db=monahrms1,pheight = ph1,compound=compound,rtime = rt, sn=sn)
}
for(i in c(1:10)){
simmzml(name=paste0('control/control',i),db=monahrms1,pheight = ph2,compound=compound,rtime = rt, sn=sn)
}
```
Then you could find 10 mzML files in case sub folder and another 10 mzML files in control sub folder, as well as corresponding csv files with m/z, retention time and compound name of the peaks.
If you prefer to simulate retention time shifts for different samples, you could add a random number to the retention time of each compound.
```{r eval=FALSE}
rt0 <- rt
set.seed(42)
for(i in c(1:10)){
rt <- rt0 + rnorm(100)
simmzml(name=paste0('case/case',i),db=monahrms1,pwidth = pw1,compound=compound,rtime = rt, sn=sn)
}
```
### Chromatography peaks
You could also use `simmzml` to stimulate tailing/leading peaks by defining the tailing factor of the peaks. When the tailing factor is lower than 1, the peaks are leading peaks. When the tailing factor is larger than 1, the peaks are tailing peaks.
```{r eval=FALSE}
# leading peaks
simmzml(name='test',db=monahrms1,pwidth = 10,compound=1,rtime = 100, sn=10,tailingfactor = 0.8)
# tailing peaks
simmzml(name='test',db=monahrms1,pwidth = 10,compound=1,rtime = 100, sn=10,tailingfactor = 1.5)
```
### matrix stimulation
You could also input a m/z vector as matrix masses. Those masses will generate background baseline signals. By default, the mass vector is from matrix samples previous [published](https://jcheminf.biomedcentral.com/articles/10.1186/s13321-022-00586-8).
```{r eval=FALSE}
data(mzm)
simmzml(name='test',db=monahrms1,pwidth = 10,compound=1,rtime = 100, sn=10,matrixmz = mzm,matrix = TRUE)
```
### Retention time simulation
We included column types and experimental retention time (in seconds) for LC-MS data and retention index for GC-MS data. You could use the following code to access such information.
```{r}
data(monahrms1)
# select compound by their index
compound <- c(1,2,3)
column <- sapply(monahrms1[compound],function(x) x$column)
# print column
column
retentiontime <- sapply(monahrms1[compound],function(x) x$rt)
# print retention time
retentiontime
data("hmdbcms")
# select compound by their index
compound <- c(1,2,300)
retentionindex <- sapply(hmdbcms[compound],function(x) x$rti)
# print retention index
retentionindex
```
## Peaks list simulation
You could also use `mzrtsim` to make simulation of peak list.
Here we make a simulation of 100 compounds from selected database with two conditions and three batches. 5 percentage of the peaks were influenced by conditions and 10 percentage of the peaks were influenced by batch effects. Three different type could be simulated: monotonic, random and block. You could also bind batch type, for example, 'mb' means the simulation would contain both monotonic and block batch effects. 'db' means the spectra database to be used for simulation as metioned in raw data simulation section.
```{r mzrtsim}
library(mzrtsim)
data("monams1")
simdata <- mzrtsim(ncomp = 100, ncond = 2, ncpeaks = 0.05,
nbatch = 3, nbpeaks = 0.1, npercond = 10, nperbatch = c(8, 5, 7), seed = 42, batchtype = 'mb', db=monams1)
```
You could save the simulated data into multiple csv files by `simdata` function. `simraw.csv` could be used for metaboanalyst. `simraw2.csv` show the raw peaks list. `simcon.csv` show peaks influenced by conditions only.`simbatchmatrix.csv` show peaks influnced by batch effects only. `simbat.csv` show peaks influenced by batch effects and conditions. `simcomp.csv` show independent peaks influenced by conditions and batch effects. `simcompchange.csv` show the conditions changes of each groups. `simblockbatchange.csv` show the block batch changes of each groups. `simmonobatchange.csv` show the monotonic batch changes of each group.
```{r simdata, eval=F}
simdata(sim,name = "sim")
```