https://github.com/jsxlei/SCALE
Single-cell ATAC-seq analysis via Latent feature Extraction
https://github.com/jsxlei/SCALE
Last synced: about 2 months ago
JSON representation
Single-cell ATAC-seq analysis via Latent feature Extraction
- Host: GitHub
- URL: https://github.com/jsxlei/SCALE
- Owner: jsxlei
- License: mit
- Created: 2018-10-19T07:22:53.000Z (over 6 years ago)
- Default Branch: master
- Last Pushed: 2023-08-23T12:06:53.000Z (almost 2 years ago)
- Last Synced: 2025-04-17T01:57:16.227Z (2 months ago)
- Language: Python
- Homepage:
- Size: 11.9 MB
- Stars: 102
- Watchers: 6
- Forks: 20
- Open Issues: 20
-
Metadata Files:
- Readme: README.md
- License: LICENSE
Awesome Lists containing this project
- awesome-atac-analysis - SCALE
README
[](https://github.com/jsxlei/scale/stargazers)
[](https://pypi.org/project/scale)
[](https://pepy.tech/project/scale-atac)
# Single-Cell ATAC-seq analysis via Latent feature Extraction
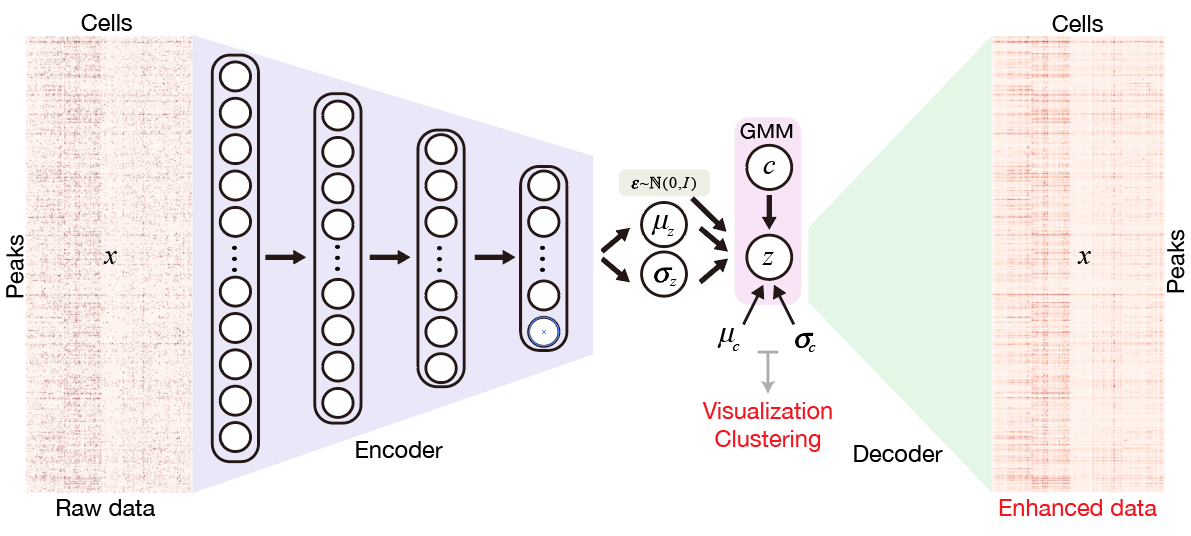
## News
2022.06.30 Introduce the highly_variable_genes from scanpy to filter peaks and support for input from multiomics data h5mu
2021.04 A new online integration tool [SCALEX](https://github.com/jsxlei/SCALEX) on scRNA-seq and scATAC-seq is available!
2021.01.14 Update to compatible with [h5ad](https://anndata.readthedocs.io/en/latest/anndata.AnnData.html) file and [scanpy](https://scanpy.readthedocs.io/en/stable/index.html)
## Installation
SCALE neural network is implemented in [Pytorch](https://pytorch.org/) framework.
Running SCALE on CUDA is recommended if available.
#### install from PyPI
pip install scale
#### install latest develop version from GitHub
pip install git+https://github.com/jsxlei/SCALE.git
or download and install
git clone git://github.com/jsxlei/SCALE.git
cd SCALE
python setup.py install
Installation only requires a few minutes.
## Quick Start
#### Input
* h5ad file
* **count matrix file**:
* row is peak and column is barcode, in **txt** / **tsv** (sep=**"\t"**) or **csv** (sep=**","**) format
* mtx **folder** contains **three files**:
* **count file**: count in **mtx** format, filename contains key word **"count"** / **"matrix"**
* **peak file**: 1-column of peaks **chr_start_end**, filename contains key word **"peak"**
* **barcode file**: 1-column of barcodes, filename contains key word **"barcode"**
* h5mu file, e.g. filename.h5mu/atac
#### Run
SCALE.py -d [input]
#### Output
Output will be saved in the output folder including:
* **model.pt**: saved model to reproduce results cooperated with option --pretrain
* **adata.h5ad**: saved data including Leiden cluster assignment, latent feature matrix and UMAP results.
* **umap.pdf**: visualization of 2d UMAP embeddings of each cell
#### Imputation
Get binary imputed data in adata.h5ad file using scanpy **adata.obsm['binary']** with option **--binary** (recommended for saving storage)
SCALE.py -d [input] --binary
or get numerical imputed data in adata.h5ad file using scanpy **adata.obsm['imputed']** with option **--impute**
SCALE.py -d [input] --impute
#### Useful options
* [--outdir] or [-o]: save results in a specific folder
* [--embed]: tSNE/UMAP, embed feature by tSNE or UMAP
* [--min_peaks]: filter low quality cells by valid peaks number, default 100
* [--min_cells]: filter low quality peaks by valid cells number, default 3 (previous default is 0.01), now replaced by [--n_feature]
* [--n_feature]: filter peaks by selecting highly variable features, default 100,000; use [--n_feature] -1 to disable.
* [--lr]: modify the initial learning rate, default is 0.002:
* [--max_iter] or [-i]: max iteration number, default is 30000
* [--seed]: random seed for parameter initialization, default is 18
* [--binary]: binarize the imputation values
* [-k]: if cluster number is known
#### Help
Look for more usage of SCALE
SCALE.py --help
Use functions in SCALE packages.
import scale
from scale import *
from scale.plot import *
from scale.utils import *
#### Running time
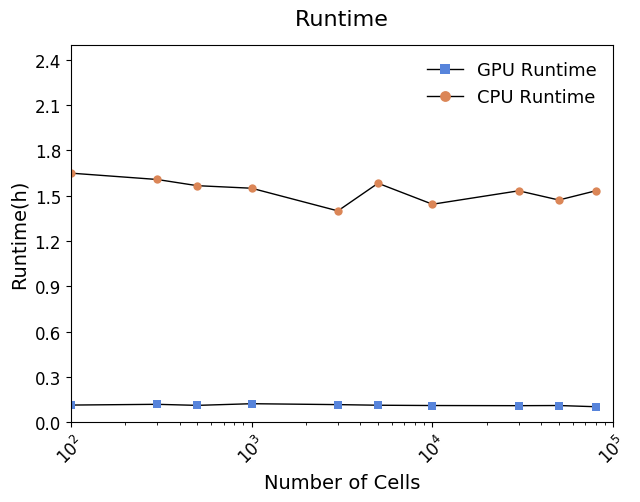
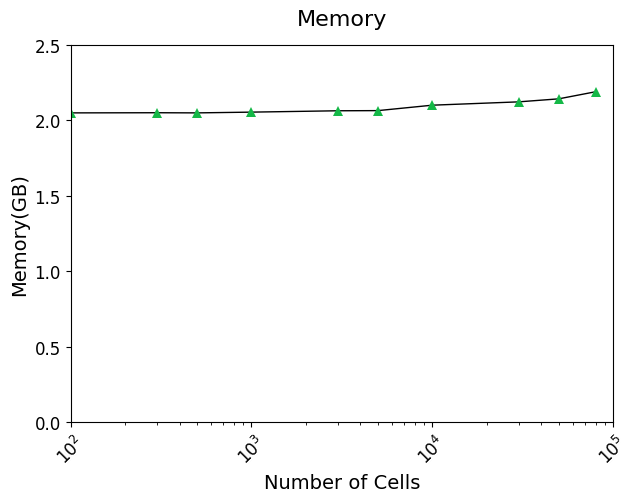
## Tutorial
**[Tutorial Forebrain](https://github.com/jsxlei/SCALE/wiki/Forebrain)** Run SCALE on dense matrix **Forebrain** dataset (k=8, 2088 cells)
#### Data availability
* [Forebrain](http://zhanglab.net/SCALE_SOURCE_DATA/Forebrain.h5ad)
* [Splenocyte](http://zhanglab.net/SCALE_SOURCE_DATA/Splenocyte.h5ad)
* [mouse_atlas](http://zhanglab.net/SCALE_SOURCE_DATA/mouse_atlas.h5ad)
* [InSilico](http://zhanglab.net/SCALE_SOURCE_DATA/InSilico.h5ad)
* [Leukemia](http://zhanglab.net/SCALE_SOURCE_DATA/Leukemia.h5ad)
* [GM12878vsHEK](http://zhanglab.net/SCALE_SOURCE_DATA/GM12878vsHEK.h5ad)
* [GM12878vsHL](http://zhanglab.net/SCALE_SOURCE_DATA/GM12878vsHL.h5ad)
* [Breast_Tumor](http://zhanglab.net/SCALE_SOURCE_DATA/Breast_Tumor.h5ad)
## Reference
[Lei Xiong, Kui Xu, Kang Tian, Yanqiu Shao, Lei Tang, Ge Gao, Michael Zhang, Tao Jiang & Qiangfeng Cliff Zhang. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nature Communications, (2019).](https://www.nature.com/articles/s41467-019-12630-7)