https://github.com/slowkow/awesome-vdj
📚 Tools and databases for analyzing HLA and VDJ genes.
https://github.com/slowkow/awesome-vdj
List: awesome-vdj
antibody awesome-list b-cell-receptor bcr bioinformatics hla hla-database hla-typing immunology t-cell-receptor tcr vdj
Last synced: 2 months ago
JSON representation
📚 Tools and databases for analyzing HLA and VDJ genes.
- Host: GitHub
- URL: https://github.com/slowkow/awesome-vdj
- Owner: slowkow
- License: cc0-1.0
- Created: 2020-01-08T19:06:40.000Z (over 5 years ago)
- Default Branch: master
- Last Pushed: 2024-05-09T17:44:03.000Z (about 1 year ago)
- Last Synced: 2024-10-21T00:34:24.349Z (8 months ago)
- Topics: antibody, awesome-list, b-cell-receptor, bcr, bioinformatics, hla, hla-database, hla-typing, immunology, t-cell-receptor, tcr, vdj
- Homepage:
- Size: 506 KB
- Stars: 204
- Watchers: 15
- Forks: 25
- Open Issues: 0
-
Metadata Files:
- Readme: README.md
- Contributing: CONTRIBUTING.md
- License: LICENSE
Awesome Lists containing this project
- ultimate-awesome - awesome-vdj - 📚 Tools and databases for analyzing HLA and VDJ genes. (Other Lists / Julia Lists)
README
# awesome-vdj 
[Antigen] presentation and recognition is central to immunology. [HLA genes] encode the proteins that present antigens. [VDJ genes][vdj] encode the receptors: T cell receptors (TCRs) in T cells and the repertoires of antibodies/immunoglobulins in B cells.
Here, researchers can find links to tools and resources for computational analysis of HLA and VDJ data.
[Contributions are welcome!](https://github.com/slowkow/awesome-vdj/blob/master/CONTRIBUTING.md)
[Antigen]: https://en.wikipedia.org/wiki/Antigen
[vdj]: https://en.wikipedia.org/wiki/V(D)J_recombination
[HLA genes]: https://en.wikipedia.org/wiki/Human_leukocyte_antigen
There are no broken links on this page if this badge is green:
[](https://github.com/slowkow/awesome-vdj/actions)
**Table of Contents**
- [VDJ Databases](#vdj-databases)
- [VDJ Analysis](#vdj-analysis)
- [HLA Databases](#hla-databases)
- [HLA Analysis](#hla-analysis)
**Related Work**
- Ming Tang's list of links here: [TCR-BCR-seq-analysis](https://github.com/crazyhottommy/TCR-BCR-seq-analysis)
---
## Literature

La Gruta, N. L., Gras, S., Daley, S. R., Thomas, P. G. & Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 18, 467–478 (2018)

Robson, K. J., Ooi, J. D., Holdsworth, S. R., Rossjohn, J. & Kitching, A. R. HLA and kidney disease: from associations to mechanisms. Nat. Rev. Nephrol. 14, 636–655 (2018)

Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 17, 281–294 (2017)
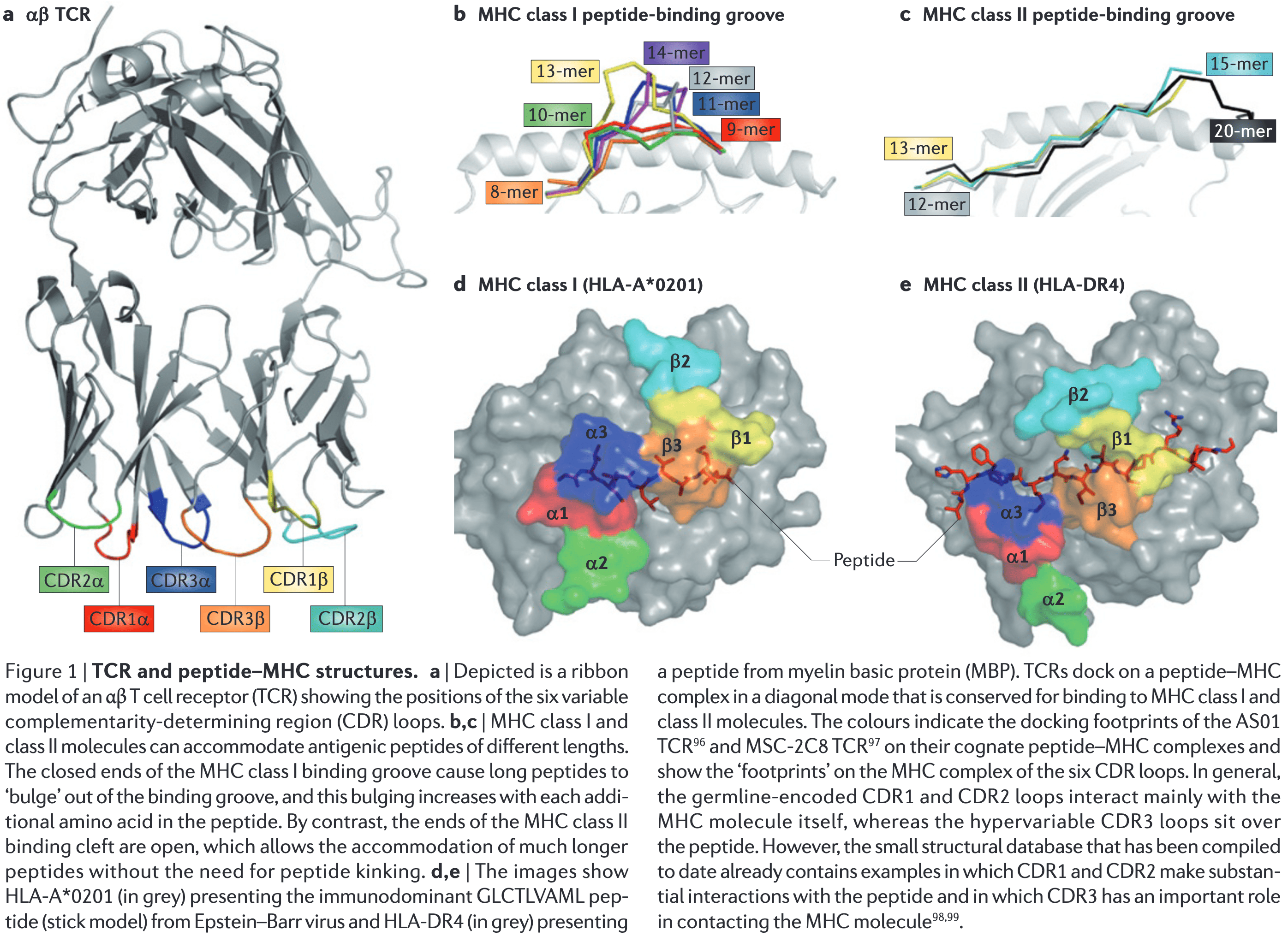
Sewell AK. Why must T cells be cross-reactive? Nat. Rev. Immunol. 12, 669–677 (2012)
- Pai, Joy A., and Ansuman T. Satpathy. [High-Throughput and Single-Cell T Cell Receptor Sequencing Technologies.](https://doi.org/10.1038/s41592-021-01201-8) Nature Methods, July 2021.
- Nordin, J., Ameur, A., Lindblad-Toh, K., Gyllensten, U., & Meadows, J. [SweHLA: the high confidence HLA typing bio-resource drawn from 1000 Swedish genomes.](https://www.nature.com/articles/s41431-019-0559-2) EJHG, 28(5), 627–635. (2020)
- Greiff, V., Yaari, G. & Cowell, L. G. [Mining adaptive immune receptor repertoires for biological and clinical information using machine learning.](http://dx.doi.org/10.1016/j.coisb.2020.10.010) Current Opinion in Systems Biology 24, 109–119 (2020)
---
## VDJ Databases
### Adaptive Immune Receptor Repertoire (AIRR) Community
http://airr-community.org
https://docs.airr-community.org/en/stable/index.html
https://github.com/airr-community
> The Adaptive Immune Receptor Repertoire (AIRR) Community of The Antibody Society is a research-driven group that is organizing and coordinating stakeholders in the use of next-generation sequencing technologies to study antibody/B-cell and T-cell receptor repertoires. Recent advances in sequencing technology have made it possible to sample the immune repertoire in exquisite detail. AIRR sequencing (AIRR-seq) has enormous promise for understanding the dynamics of the immune repertoire in vaccinology, infectious diseases, autoimmunity, and cancer biology, but also poses substantial challenges. The AIRR Community was established to meet these challenges.
### A Public Database of Memory and Naive B-Cell Receptor Sequences
https://datadryad.org/stash/dataset/doi:10.5061/dryad.35ks2
> We present a public database of more than 37 million unique BCR sequences from three healthy adult donors that is many fold deeper than any existing resource, together with a set of online tools designed to facilitate the visualization and analysis of the annotated data.
DeWitt, W. S. et al. [A Public Database of Memory and Naive B-Cell Receptor Sequences.](https://pubmed.ncbi.nlm.nih.gov/27513338/) PLoS One 11, e0160853 (2016)
### Coronavirus-Binding Antibody Sequences & Structures
http://opig.stats.ox.ac.uk/webapps/covabdab/
> The Oxford Protein Informatics Group (Dept. of Statistics, University of Oxford) is collaborating in efforts to understand the immune response to SARS-CoV2 infection and vaccination. As part of our investigations, we are releasing and maintaining this public database to document all published/patented binding antibodies and nanobodies to coronaviruses, including SARS-CoV2, SARS-CoV1, and MERS-CoV.
### Human Vaccines Project (Human Immunome Program)
https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP174305
> The Human Immunome Program (HIP) is open-source effort with the goal sequencing all of the adaptive receptors on the surface of human B and T cells. Under a targeted 7-to-10-year effort, the program will sequence these receptors from a group of globally diverse individuals, and determine the structure and function of a key subset of receptors. Through an open-source approach, this data will be made available to researchers across the world.
### immuneACCESS
https://clients.adaptivebiotech.com/immuneaccess
> Dive into the world’s largest collection of TCR and BCR sequences. Easily incorporate millions of sequences worth of public data into your next papers and projects using immunoSEQ Analyzer. Construct your own projects, draw your own conclusions, and freely publish new discoveries.
See [these instructions](https://github.com/slowkow/awesome-vdj/blob/master/download-from-immuneaccess.md) for tips on how to download data from immuneACCESS.
### iReceptor
https://gateway.ireceptor.org/home
> iReceptor facilitates the curation, analysis and sharing of antibody/B-cell and T-cell receptor repertoires (Adaptive Immune Receptor Repertoire or AIRR-seq data) from multiple labs and institutions. We are committed to providing a platform for researchers to increase the value of their data through sharing with the community. This will greatly increase the amount of data available to answer complex questions about the adaptive immune response, accelerating the development of vaccines, therapeutic antibodies against autoimmune diseases, and cancer immunotherapies.
### McPAS-TCR: A manually curated catalogue of pathology associated T-cell receptor sequences
https://friedmanlab.weizmann.ac.il/McPAS-TCR/
> McPAS-TCR is a manually curated catalogue of T cell receptor (TCR) sequences that were found in T cells associated with various pathological conditions in humans and in mice. It is meant to link TCR sequences to their antigen target or to the pathology and organ with which they are associated.
### PIRD: Pan immune repertoire database
https://db.cngb.org/pird/
> Pan immune repertoire database (PIRD) collects raw and processed sequences of immunoglobulins (IGs) and T cell receptors (TCRs) of human and other vertebrate species with different phenotypes. You can check the detailed information of each sample in the database, choose samples to analyze according to your need, and upload data to analyze. Your analysis results will be auto-saved, so you can return to check them at any time. PIRD is developed by the immune and health lab of BGI-research.
Zhang, W. et al. [PIRD: Pan Immune Repertoire Database.](https://doi.org/10.1093/bioinformatics/btz614) Bioinformatics 36, 897–903 (2020)
### STCRDab: The Structural T-Cell Receptor Database
http://opig.stats.ox.ac.uk/webapps/stcrdab/
> An automated, curated set of T-Cell Receptor structural data from the PDB.
### TCR3d: T cell receptor structural repertoire database
https://tcr3d.ibbr.umd.edu/
> Welcome to the T cell receptor (TCR) structural repertoire database. Here we provide an easy-to-use interface to view all experimentally determined T cell receptor structures and their complexes. This includes complementarity determining region loops and analysis of interfaces with antigenic peptide and MHC.
>
> We have also assembled a set of known TCR sequences from recent studies including TCR repertoire sequencing efforts.
>
> The major goal of this site is to enable insights into the basis of TCR structure and recognition, to assist efforts in predictive modeling of this key component of the adaptive immune response, and to facilitate rational engineering of improved and novel immunotherapeutics.
### VDJDB: A curated database of T-cell receptor sequences of known antigen specificity
https://vdjdb.cdr3.net
https://github.com/antigenomics/vdjdb-db
> The primary goal of VDJdb is to facilitate access to existing information on T-cell receptor antigen specificities, i.e. the ability to recognize certain epitopes in certain MHC contexts.
>
> Our mission is to both aggregate the scarce TCR specificity information available so far and to create a curated repository to store such data.
---
## VDJ Analysis
### Absolut: Unconstrained lattice antibody-antigen bindings generator - One tool to simulate them all!
C++ software
https://github.com/csi-greifflab/Absolut
> Absolut! is a database and C++ user interface that allows the high-throughput computation for the 3D-lattice binding of any CDRH3 sequence to any antigen, enabling the custom generation of new antibody-antigen structural datasets for ML training or testing.
Robert, P. A. et al. [A billion synthetic 3D-antibody-antigen complexes enable unconstrained machine-learning formalized investigation of antibody specificity prediction.](https://doi.org/10.1101/2021.07.06.451258) bioRxiv 2021.07.06.451258 (2021)
### ALICE: Antigen-specific Lymphocyte Identification by Clustering of Expanded sequences
R scripts
https://github.com/pogorely/ALICE
> Detecting TCR involved in immune responses from single RepSeq datasets.
Pogorelyy, M. V. et al. [Detecting T cell receptors involved in immune responses from single repertoire snapshots.](https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000314) PLoS Biol. 17, e3000314 (2019)
### CONGA: Clonotype Neighbor Graph Analysis
Python scripts and C++ code
https://github.com/phbradley/conga
> CONGA was developed to detect correlation between T cell gene expression profile and TCR sequence in single-cell datasets.
Schattgen, S. A. et al. [Linking T cell receptor sequence to transcriptional profiles with clonotype neighbor graph analysis (CoNGA).](https://www.biorxiv.org/content/10.1101/2020.06.04.134536v1) Cold Spring Harbor Laboratory 2020.06.04.134536 (2020) doi:10.1101/2020.06.04.134536
### ClusTCR: a Python interface for rapid clustering of large sets of CDR3 sequences with unknown antigen specificity
Python package
https://github.com/svalkiers/clusTCR
> CDR3 clustering module providing a new method for fast and accurate clustering of large data sets of CDR3 amino acid sequences, and offering functionalities for downstream analysis of clustering results.
>
> A two-step clustering approach that combines the speed of the Faiss Clustering Library with the accuracy of Markov Clustering Algorithm
> On a standard machine*, clusTCR can cluster 1 million CDR3 sequences in under 5 minutes.
### DeepTCR: Deep Learning Methods for Parsing T-Cell Receptor Sequencing (TCRSeq) Data
Python package
https://github.com/sidhomj/DeepTCR
> DeepTCR is a python package that has a collection of unsupervised and supervised deep learning methods to parse TCRSeq data. It has the added functionality of being able to analyze paired alpha/beta chain inputs as well as also being able to take in v/d/j gene usage and the contextual HLA information the TCR-Sequences were seen in (i.e. HLA alleles for a repertoire from a given human sample).
### dkm: Dynamic Kernel Matching
Python scripts
https://github.com/jostmey/dkm
> DKM is analogous to a convolutional network, but for sequences. Consider the problem of classifying a sequence. Because some sequences are longer than others, the number of features is irregular. Given a specific sequence, the challenge is to determine the appropriate permutation of features with weights, allowing us to run the features through the statistical classifier to generate a prediction. To find the permutation of features that exhibit the maximal response, like how max-pooling identifies the image patch that exhibit the maximal response, we use a sequence alignment algorithm.
### enclone
Standalone binary, Linux
https://10xgenomics.github.io/enclone/
https://github.com/10XGenomics/enclone
> enclone is standalone software (primarily written in Rust) developed by 10x Genomics for analysis of single cell TCR and BCR sequences. enclone performs SHM-aware clonotyping, phylogenetic/lineage analysis, multiple sequence alignment, and provides an extremely fast interface to analyze, display, and export VDJ, gene expression, and feature barcoding (REAP-seq, CITE-seq, ECCITE-seq, LIBRA-seq, PERTURB-seq, _etc._) data.
### GIANA: Geometry Isometry based TCR AligNment Algorithm
Python scripts
https://github.com/s175573/GIANA
> Geometric Isometry-
based TCR AligNment Algorithm (GIANA), a mathematical framework to transform the CDR3 sequences, which converted the sequence alignment and clustering problem into a classic nearest neighbor search in the high-dimensional Euclidean space.
Zhang H, Zhan X, Li B. [GIANA allows computationally-efficient TCR clustering and multi-disease repertoire classification by isometric transformation.](https://www.ncbi.nlm.nih.gov/pubmed/34349111) Nat Commun. 2021;12: 4699. doi:10.1038/s41467-021-25006-7
### immunarch: An R Package for Painless Bioinformatics Analysis of T-cell and B-cell Immune Repertoire Data
R package
https://github.com/immunomind/immunarch
> immunarch is an R package designed to analyse T-cell receptor (TCR) and B-cell receptor (BCR)
> repertoires, aimed at medical scientists and bioinformaticians. The mission of immunarch is to
> make immune sequencing data analysis as effortless as possible and help you focus on research
> instead of coding. Follow us on Twitter for news and updates.
### immuneSIM: Tunable Simulation of B- And T-Cell Receptor Repertoires
R package
https://CRAN.R-project.org/package=immuneSIM
https://github.com/GreiffLab/immuneSIM
> Simulate full B-cell and T-cell receptor repertoires using an in silico recombination process
> that includes a wide variety of tunable parameters to introduce noise and biases. Additional
> post-simulation modification functions allow the user to implant motifs or codon biases as
> well as remodeling sequence similarity architecture. The output repertoires contain records
> of all relevant repertoire dimensions and can be analyzed using provided repertoire analysis
> functions. Preprint is available at bioRxiv (Weber et al., 2019 ).
Weber, C. R. et al. [immuneSIM: tunable multi-feature simulation of B- and T-cell receptor repertoires for immunoinformatics benchmarking.](https://pubmed.ncbi.nlm.nih.gov/32154832/) Bioinformatics 36, 3594–3596 (2020)
### ImReP: Rapid and accurate profiling of the adaptive immune repertoires from regular RNA-Seq data
Python scripts
https://github.com/Mangul-Lab-USC/imrep
> ImReP is a method to quantify individual immune response based on a recombination landscape of genes encoding B and T cell receptors (BCR and TCR). ImReP is able to efficiently extract TCR and BCR reads from the RNA-Seq data and assemble clonotypes (defined as clones with identical CDR3 amino acid sequences) and detect corresponding V(D)J recombinations. Using CAST clustering technique, ImReP is able to correct assembled clonotypes for PCR and sequencing errors.
Mandric, I., Rotman, J., Yang, H.T. et al. [Profiling immunoglobulin repertoires across multiple human tissues using RNA sequencing.](https://doi.org/10.1038/s41467-020-16857-7) Nat Commun 11, 3126 (2020).
### IMSEQ: IMmunogenetic SEQuence Analysis
C++ tools
https://github.com/lkuchenb/imseq
> IMSEQ is a fast, PCR and sequencing error aware tool to analyze high throughput data from recombined T-cell receptor or immunoglobolin gene sequencing experiments. It derives immune repertoires from sequencing data in FASTA / FASTQ format.
### MiGMAP: mapper for full-length T- and B-cell repertoire sequencing
Groovy and Java tools
https://github.com/mikessh/migmap
> In a nutshell, this software is a smart wrapper for IgBlast V-(D)-J mapping tool designed to facilitate analysis immune receptor libraries profiled using high-throughput sequencing. This package includes additional experimental modules for contig assembly, error correction and immunoglobulin lineage tree construction.
### MiXCR: a universal tool for fast and accurate analysis of T- and B- cell receptor repertoire sequencing data
Java tools
https://github.com/milaboratory/mixcr
> MiXCR is a universal framework that processes big immunome data from raw sequences to quantitated clonotypes. MiXCR efficiently handles paired- and single-end reads, considers sequence quality, corrects PCR errors and identifies germline hypermutations. The software supports both partial- and full-length profiling and employs all available RNA or DNA information, including sequences upstream of V and downstream of J gene segments.
### mvTCR
Python scripts and Jupyter notebooks
https://github.com/SchubertLab/mvTCR
Trained models: https://zenodo.org/record/5006839
> A multi-view Variational Autoencoder (mvTCR) to jointly embed transcriptomic and TCR sequence information at a single-cell level to better capture the phenotypic behavior of T cells.
An Y, Drost F, Theis F, Schubert B, Lotfollahi M. [Jointly learning T-cell receptor and transcriptomic information to decipher the immune response.](https://www.biorxiv.org/content/10.1101/2021.06.24.449733v1) bioRxiv. 2021. p. 2021.06.24.449733.
### PRESTO: The REpertoire Sequencing TOolkit
Python package
https://presto.readthedocs.io/en/stable
https://bitbucket.org/kleinstein/presto
> pRESTO is a toolkit for processing raw reads from high-throughput sequencing of B cell and T cell repertoires.
>
> The REpertoire Sequencing TOolkit (pRESTO) is composed of a suite of utilities to handle all stages of sequence processing prior to germline segment assignment. pRESTO is designed to handle either single reads or paired-end reads. It includes features for quality control, primer masking, annotation of reads with sequence embedded barcodes, generation of unique molecular identifier (UMI) consensus sequences, assembly of paired-end reads and identification of duplicate sequences. Numerous options for sequence sorting, sampling and conversion operations are also included.
Heiden, J. A. V. et al. [pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires.](https://pubmed.ncbi.nlm.nih.gov/24618469/) Bioinformatics 30, 1930–1932 (2014)
### pyIR: An IgBLAST wrapper and parser
Python package
https://github.com/crowelab/PyIR
> PyIR is a minimally-dependent high-speed wrapper for the IgBLAST immunoglobulin and T-cell analyzer. This is achieved through chunking the input data set and running IgBLAST single-core in parallel to better utilize modern multi-core and hyperthreaded processors.
### Recon: Reconstruction of Estimated Communities from Observed Numbers
Python scripts
https://github.com/ArnaoutLab/Recon
> Recon uses the distribution of species counts in a sample to estimate the distribution of species counts in the population from which the sample was drawn.
Kaplinsky, J. & Arnaout, R. [Robust estimates of overall immune-repertoire diversity from high-throughput measurements on samples.](https://pubmed.ncbi.nlm.nih.gov/27302887/) Nat. Commun. 7, 11881 (2016)
### scirpy: A scanpy extension to analyse single-cell TCR data.
Python package
https://github.com/icbi-lab/scirpy
> Scirpy is a scalable python-toolkit to analyse T cell receptor (TCR) repertoires from single-cell RNA sequencing (scRNA-seq) data. It seamlessly integrates with the popular scanpy library and provides various modules for data import, analysis and visualization.
### scRepertoire: A toolkit for single-cell immune profiling
R package
https://github.com/ncborcherding/scRepertoire
> scRepertoire v1.0.0 added the functionality of the powerTCR approach to comparing clone size distribution, please cite the manuscript if using the clonesizeDistribution() function. Similiarly, the application of novel indices for single-cell clonotype dynamics in the StartracDiversity() function is based on the work from Lei Zhang et al.
Borcherding, N., Bormann, N. L. & Kraus, G. [scRepertoire: An R-based toolkit for single-cell immune receptor analysis.](https://pubmed.ncbi.nlm.nih.gov/32789006/) F1000Res. 9, 47 (2020)
### tcr-bert
Python scripts
https://github.com/wukevin/tcr-bert
Categories: embedding, predicting antigens
> TCR-BERT is a large language model trained on T-cell receptor sequences, built using a lightly modified BERT architecture with tweaked pre-training objectives.
Wu K, Yost KE, Daniel B, Belk JA, Xia Y, Egawa T, et al. [TCR-BERT: learning the grammar of T-cell receptors for flexible antigen-xbinding analyses.](http://dx.doi.org/10.1101/2021.11.18.469186) bioRxiv. 2021. p. 2021.11.18.469186. doi:10.1101/2021.11.18.469186
### TCRconv
Python package
https://github.com/emmijokinen/TCRconv
> TCRconv is a deep learning model for predicting recognition between T cell receptors and epitopes. It uses protBERT embeddings for the TCRs and convolutional neural networks for the prediction.
Jokinen E, Dumitrescu A, Huuhtanen J, Gligorijević V, Mustjoki S, Bonneau R, et al. [TCRconv: predicting recognition between T cell receptors and epitopes using contextualized motifs.](https://pubmed.ncbi.nlm.nih.gov/36477794/) Bioinformatics. 2023;39. doi:10.1093/bioinformatics/btac788
### TCRGP
Python scripts
https://github.com/emmijokinen/TCRGP
> TCRGP is a novel Gaussian process method that can predict if TCRs recognize certain epitopes. This method can utilize different CDR sequences from both TCRα and TCRβ chains from single-cell data and learn which CDRs are important in recognizing the different epitopes. TCRGP has been developed at Aalto University.
Jokinen E, Huuhtanen J, Mustjoki S, Heinonen M, Lähdesmäki H. [Predicting recognition between T cell receptors and epitopes with TCRGP.](https://pubmed.ncbi.nlm.nih.gov/33764977/) PLoS Comput Biol. 2021;17: e1008814.
### tcr-dist
Python scripts
https://github.com/phbradley/tcr-dist
> Software tools for the analysis of epitope-specific T cell receptor (TCR) repertoires
Dash P, Fiore-Gartland A, Hertz T et al. [Quantifiable predictive features define epitope-specific T cell receptor repertoires.](https://pubmed.ncbi.nlm.nih.gov/28636592/) Nature. 2017;547(7661): 89-93.
### tcrdist3
Python package
https://github.com/kmayerb/tcrdist3
> tcrdist3 is a Python API-enabled toolkit for analyzing T-cell receptor repertoires. Some of the functionality and code is adapted from the original tcr-dist package.
Mayer-Blackwell K, Schattgen S, Cohen-Lavi L et al. [TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs.](https://pubmed.ncbi.nlm.nih.gov/34845983/) eLife. 2021;10: e68605.
### TCRdock
Python scripts
https://github.com/phbradley/TCRdock
> Python tools for TCR:peptide-MHC modeling and analysis:
> - Set up and run TCR-specialized AlphaFold simulations starting from a TSV file with TCR, peptide, and MHC information.
> - Parse a TCR:peptide-MHC ternary PDB structure and define V/J/CDR3, MHC allele, TCR and MHC coordinate frames, and TCR:pMHC docking geometry
> - Calculate distances between docking geometries ('docking RMSDs') for use in clustering/docking analysis and model evaluation.
Bradley P. [Structure-based prediction of T cell receptor:peptide-MHC interactions.](https://pubmed.ncbi.nlm.nih.gov/36661395/) Elife. 2023;12: e82813. doi:10.7554/eLife.82813
### TCRmodel2: high-resolution modeling of T cell receptor recognition using deep learning
Python scripts
https://github.com/piercelab/tcrmodel2
> This method, named TCRmodel2, allows users to submit sequences through an easy-to-use interface and shows similar or greater accuracy than AlphaFold and other methods to model TCR–peptide–MHC complexes based on benchmarking. It can generate models of complexes in 15 minutes, and output models are provided with confidence scores and an integrated molecular viewer
Yin R, Ribeiro-Filho HV, Lin V, Gowthaman R, Cheung M, Pierce BG. [TCRmodel2: high-resolution modeling of T cell receptor recognition using deep learning.](https://www.ncbi.nlm.nih.gov/pubmed/37140040) Nucleic Acids Res. 2023;51: W569–W576. doi:10.1093/nar/gkad356
### TITAN - Tcr epITope bimodal Attention Networks
Python package
https://github.com/PaccMann/TITAN
> a bimodal neural network that explicitly encodes both TCR sequences and epitopes to enable the independent study of generalization capabilities to unseen TCRs and/or epitopes.
Weber A, Born J, Rodriguez Martínez M. TITAN: [T-cell receptor specificity prediction with bimodal attention networks.](https://pubmed.ncbi.nlm.nih.gov/34252922/) Bioinformatics. 2021;37: i237–i244.
### TRUST4: TCR and BCR assembly from RNA-seq data
C/C++ code
https://github.com/liulab-dfci/TRUST4
> Tcr Receptor Utilities for Solid Tissue (TRUST) is a computational tool to analyze TCR and BCR sequences using unselected RNA sequencing data, profiled from solid tissues, including tumors. TRUST4 performs de novo assembly on V, J, C genes including the hypervariable complementarity-determining region 3 (CDR3) and reports consensus of BCR/TCR sequences. TRUST4 then realigns the contigs to IMGT reference gene sequences to report the corresponding information. TRUST4 supports both single-end and paired-end sequencing data with any read length.
Song L, Cohen D, Ouyang Z, Cao Y, Hu X, Shirley Liu X. [TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data.](https://www.nature.com/articles/s41592-021-01142-2) Nat Methods. Nature Publishing Group; 2021 May 13;1–4.
### vampire: Deep generative models for TCR sequences
Python package
https://github.com/matsengrp/vampire/
> Fit and test variational autoencoder (VAE) models for T cell receptor sequences.
Davidsen, K. et al. [Deep generative models for T cell receptor protein sequences.](http://dx.doi.org/10.7554/eLife.46935) Elife 8, e46935 (2019)
### VDJtools
Groovy and Java tools
https://github.com/mikessh/vdjtools
> A comprehensive analysis framework for T-cell and B-cell repertoire sequencing data
---
## HLA Databases
### Nomenclature of HLA Alleles
https://hla.alleles.org/nomenclature/index.html

### Allele Frequency Net Database
http://www.allelefrequencies.net/
> AFND is a public resource that collects information on allele, genotype and haplotype frequencies from different polymorphic areas in the human genome such as human leukocyte antigens (HLA), killer-cell immunoglobulin-like receptors, etc. To produce this database we have compiled a large collection of datasets from different sources including: (i) peer-reviewed literature, (ii) datasets from international workshops in immunogenetics and histocompatiblity and (iii) data submitted directly to AFND by individual laboratories. As more than 75% of the submissions in AFND are derived from peer-review literature, we rely upon data verification by journal editors and reviewers when source studies are published.
Gonzalez-Galarza FF, McCabe A, Santos EJMD, Jones J, Takeshita L, Ortega-Rivera ND, et al. [Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools.](https://pubmed.ncbi.nlm.nih.gov/31722398/) Nucleic Acids Res. 2020;48: D783–D788. doi:10.1093/nar/gkz1029
### IEDB: Immune Epitope Database and Analysis Resource
https://www.iedb.org/
> The Immune Epitope Database (IEDB) is a freely available resource funded by NIAID. It catalogs experimental data on antibody and T cell epitopes studied in humans, non-human primates, and other animal species in the context of infectious disease, allergy, autoimmunity and transplantation. The IEDB also hosts tools to assist in the prediction and analysis of epitopes.
### IMGTHLA
https://www.ebi.ac.uk/ipd/imgt/hla/
https://github.com/ANHIG/IMGTHLA
> The IPD-IMGT/HLA Database provides a specialist database for sequences of the human major histocompatibility complex (MHC) and includes the official sequences named by the WHO Nomenclature Committee For Factors of the HLA System. The IPD-IMGT/HLA Database is part of the international ImMunoGeneTics project (IMGT).
>
> The database uses the 2010 naming convention for HLA alleles in all tools herein. To aid in the adoption of the new nomenclature, all search tools can be used with both the current and pre-2010 allele designations. The pre-2010 nomenclature designations are only used where older reports or outputs have been made available for download.
---
## HLA Analysis
### arcasHLA: Fast and accurate in silico inference of HLA genotypes from RNA-seq
Python scripts
https://github.com/RabadanLab/arcasHLA
> arcasHLA performs high resolution genotyping for HLA class I and class II genes from RNA sequencing, supporting both paired and single-end samples.
### BIGDAWG: Case-Control Analysis of Multi-Allelic Loci
R package
https://github.com/IgDAWG/BIGDAWG
> Data sets and functions for chi-squared Hardy-Weinberg and case-control association tests of highly polymorphic genetic data [e.g., human leukocyte antigen (HLA) data]. Performs association tests at multiple levels of polymorphism (haplotype, locus and HLA amino-acids) as described in Pappas DJ, Marin W, Hollenbach JA, Mack SJ (2016) doi:10.1016/j.humimm.2015.12.006. Combines rare variants to a common class to account for sparse cells in tables as described by Hollenbach JA, Mack SJ, Thomson G, Gourraud PA (2012) doi:10.1007/978-1-61779-842-9_14.
### HATK: HLA Analysis Toolkit
Python scripts
https://github.com/WansonChoi/HATK
> HATK(HLA Analysis Tool-Kit) is a collection of tools and modules to perform HLA fine-mapping analysis, which is to identify which HLA allele or amino acid position of the HLA gene is driving the disease.
### hla3: weight of evidence of HLA allele expression based on bulk TCR beta-chain repertoires
Python scripts
https://github.com/kmayerb/hla3
> This repository contains Python functions for inferring HLA-alleles from bulk TCR beta chain data using a simple weight of evidence predictor.
### HLA-LA: Fast HLA type inference from whole-genome data
C++ and Perl
https://github.com/DiltheyLab/HLA-LA
> HLA typing based on a population reference graph and employs a new linear projection method to align reads to the graph.
### HLA-TAPAS: HLA-Typing At Protein for Association Studies
Python and R scripts
https://github.com/immunogenomics/HLA-TAPAS
> An HLA-focused pipeline that can handle HLA reference panel construction (MakeReference), HLA imputation (SNP2HLA), and HLA association (HLAassoc). It is an updated version of the SNP2HLA.
### HLAProfiler: Using k-mers to call HLA alleles in RNA sequencing data
Perl scripts
https://github.com/ExpressionAnalysis/HLAProfiler
> HLAProfiler uses the k-mer content of next generation sequencing reads to call HLA types in a sample. Based on the k-mer content each each read pair is assigned to an HLA gene and the aggregate k-mer profile for the gene is compared to reference k-mer profiles to determin the HLA type. Currently HLAProfiler only supports paired-end RNA-seq data.
### hlabud: HLA genotype analysis in R
R package
https://github.com/slowkow/hlabud
> hlabud provides methods to retrieve sequence alignment data from IMGTHLA and convert the data into convenient R matrices ready for downstream analysis. See the usage examples to learn how to use the data with logistic regression and dimensionality reduction. We also share tips on how to visualize the 3D molecular structure of HLA proteins and highlight specific amino acid residues.
### HLAtools: Functions and Datasets for HLA Informatics
R package
https://github.com/sjmack/HLAtools
> We have developed HLAtools, an R package that automates the consumption of IPD-IMGT/HLA resources, renders them computable, and makes them available alongside tools for data analysis, visualization and investigation. Tthe package is compatible with all IPD-IMGT/HLA Database release versions up to release 3.58.0.
### Kourami: Graph-guided assembly for HLA alleles
Bash scripts and Java code
https://github.com/Kingsford-Group/kourami
> Kourami is a graph-guided assembler for HLA haplotypes covering typing exons (exons 2 and 3 for Class I and exon 3 for Class II) using high-coverage whole genome sequencing data. Kourami constructs highly accurate haplotype sequences at 1-bp resolution by first encoding currently available HLA allelic sequences from IPD-IMGT/HLA Database http://www.ebi.ac.uk/ipd/imgt/hla/ as partial-ordered graphs.
Lee, H., & Kingsford, C. [Kourami: graph-guided assembly for novel human leukocyte antigen allele discovery.](https://pubmed.ncbi.nlm.nih.gov/29415772/) Genome Biology 19(16), 2018
### MATER: Minimizer RNAseq HLA typer
Python scripts and C code
https://github.com/cschin/MATER
> MATER is a minimizer-based HLA typer for RNAseq read dataset. In a typical RNAseq dataset, the reads sampled from HLA genes are less uniform and may miss regions that makes assembly or variant calling base methods for HLA typing more challenge. Here we adopt a slight different approach. We try to assign each reads to possible HLA types by using minimizers. Namely, we will generate dense minimizer for each reads and compare to those from the HLA type seqeunces.
> We annotate each each reads to possible HLA serotype or 4 digit type sequence according the minimizer matches. Some reads may be able to assign to single HLA type-sequence, some other may be more ambiguous. We derive a simple score to summarize the results from all reads that are mapped to HLA-type sequences for each HLA allele.
### MultiHLA: WES HLA Typing based on multiple alternative tools
Snakemake workflows
https://github.com/lkuchenb/MultiHLA
> This workflow enables the concurrent analysis of WES or WGS data using publicly available software to derive HLA haplotypes from this type of data.
> It includes automated Snakemake workflows for the following tools: xHLA, HLA-VBSeq, OptiType, HLA-LA, arcasHLA
### OptiType: Precision HLA typing from next-generation sequencing data
Python scripts
https://github.com/FRED-2/OptiType
> OptiType is a novel HLA genotyping algorithm based on integer linear programming, capable of producing accurate 4-digit HLA genotyping predictions from NGS data by simultaneously selecting all major and minor HLA Class I alleles.
### PHLAT: Inference of High Resolution HLA Types
Python scripts
https://sites.google.com/site/phlatfortype/home
> PHLAT is a bioinformatics algorithm that offers HLA typing at four-digit resolution (or higher) using genome-wide transcriptome and exome sequencing data over a wide range of read lengths and sequencing depths.
### seq2HLA: HLA typing from RNA-Seq sequence reads
Python scripts
https://github.com/TRON-Bioinformatics/seq2HLA
> In-silico method written in Python and R to determine HLA genotypes of a sample. seq2HLA takes standard RNA-Seq sequence reads in fastq format as input, uses a bowtie index comprising all HLA alleles and outputs the most likely HLA class I and class II genotypes (in 4 digit resolution), a p-value for each call, and the expression of each class.
### SNP2HLA: Imputation of Amino Acid Polymorphisms in Human Leukocyte Antigens
C shell script
http://software.broadinstitute.org/mpg/snp2hla/
> SNP2HLA is a tool to impute amino acid polymorphisms and single nucleotide polymorphisms in human luekocyte antigenes (HLA) within the major histocompatibility complex (MHC) region in chromosome 6.
### T1K: efficient and accurate inference of KIR or HLA alleles from RNA-seq, whole-genome sequencing, or whole-exome sequencing data
C++ tool and Perl scripts
https://github.com/mourisl/T1K
> T1K (The ONE genotyper for Kir and HLA) is a computational tool to infer the alleles for the polymorphic genes such as KIR and HLA. T1K calculates the allele abundances based on the RNA-seq/WES/WGS read alignments on the provided allele reference sequences. The abundances are used to pick the true alleles for each gene. T1K provides the post analysis steps, including novel SNP detection and single-cell representation. T1K supports both single-end and paired-end sequencing data with any read length.
Song L, Bai G, Liu XS, Li B, Li H. [Efficient and accurate KIR and HLA genotyping with massively parallel sequencing data.](https://pubmed.ncbi.nlm.nih.gov/37169596/) Genome Res. 2023 [cited 13 May 2023]. doi:10.1101/gr.277585.122
### xHLA: Fast and accurate HLA typing from short read sequence data
Bash, Perl, Python, and R scripts
https://github.com/humanlongevity/HLA
> xHLA iteratively refines the mapping results at the amino acid level to achieve 99 to 100% 4-digit typing accuracy for both class I and II HLA genes, taking only about 3 minutes to process a 30X whole genome BAM file on a desktop computer.
Xie, C., Yeo, Z. X., Wong, M., Piper, J., Long, T., Kirkness, E. F., ... & Brady, C. (2017). [Fast and accurate HLA typing from short-read next-generation sequence data with xHLA.](https://pubmed.ncbi.nlm.nih.gov/28674023) Proceedings of the National Academy of Sciences, 114(30), 8059-8064.